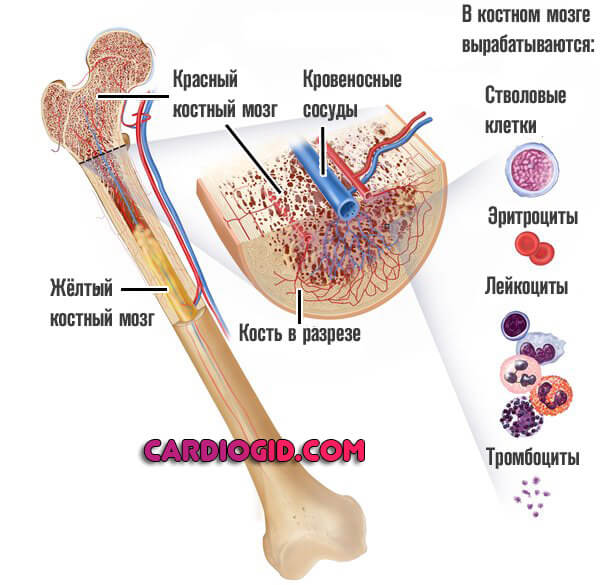
Анемии. классификация анемий
Содержание:
Лечение
При обострении ежедневно вводят витамин B12 (по 100—200 мкг) в течение 1-й недели до ретикулоцитарного криза, который обычно наступает на 5-й день от начала введения витамина B12 и характеризуется увеличенным содержанием ретикулоцитов. В дальнейшем те же дозы вводят через день в течение 4—5 недель до наступления гематол, ремиссии, которая определяется нормализацией периферической крови и костномозгового кроветворения и восстановлением нормального содержания витамина B12 в крови, причем нормализация крови начинается вслед за ретикулоцитарным кризом и завершается через 4—6 недель; нормализация костномозгового кроветворения (трансформация мегалобластиче-ского эритроцитопоэза в нормобласти-ческий) начинается немедленно после введения витамина B12 и завершается через 48—72 часа; содержание витамина B12 в крови восстанавливается постепенно. В период ремиссии исчезают клин, симптомы, включая неврологические. При фуникулярном миелозе вводят массивные дозы витамина B12 (500—1000 мкг) ежедневно в течение 7—10 дней, в дальнейшем два раза в неделю до исчезновения неврологических симптомов.
При психозах в зависимости от клин, картины применяют психотропные средства. При быстро прогрессирующей тяжелой пернициозной анемии (угрожающая Пернициозная кома) показано переливание эритроцитной массы (см. Переливание крови).
Другие заболевания из группы Болезни крови, кроветворных органов и отдельные нарушения, вовлекающие иммунный механизм:
| B12-дефицитная анемия |
| Анемии, обусловленные нарушением синтеза утилизацией порфиринов |
| Анемии, обусловленные нарушением структуры цепей глобина |
| Анемии, характеризующиеся носительством патологически нестабильных гемоглобинов |
| Анемия Фанкони |
| Анемия, связанная со свинцовым отравлением |
| Апластическая анемия |
| Аутоиммунная гемолитическая анемия |
| Аутоиммунная гемолитическая анемия с неполными тепловыми агглютининами |
| Аутоиммунная гемолитическая анемия с полными Холодовыми агглютининами |
| Аутоиммунная гемолитическая анемия с тепловыми гемолизинами |
| Болезни тяжелых цепей |
| болезнь Верльгофа |
| Болезнь Виллебранда |
| болезнь Ди Гулъелъмо |
| болезнь Кристмаса |
| Болезнь Маркиафавы-Микели |
| Болезнь Рандю — Ослера |
| Болезнь тяжелых альфа-цепей |
| Болезнь тяжелых гамма-цепей |
| Болезнь Шенлейн — Геноха |
| Внекостномозговые поражения |
| Волосатоклеточный лейкоз |
| Гемобластозы |
| Гемолитико-уремический синдром |
| Гемолитико-уремический синдром |
| Гемолитическая анемия, связанная с дефицитом витамина Е |
| Гемолитическая анемия, связанная с дефицитом глюкозо-6-фосфатдегидрогеназы (Г-6-ФДГ) |
| Гемолитическая болезнь плода и новорожденного |
| Гемолитические анемии, связанные с механическим повреждением эритроцитов |
| Геморрагическая болезнь новорожденных |
| Гистиоцитоз злокачественный |
| Гистологическая классификация лимфогранулематоза |
| ДВС-синдром |
| Дефицит К-витаминзависимых факторов |
| Дефицит фактора I |
| Дефицит фактора II |
| Дефицит фактора V |
| Дефицит фактора VII |
| Дефицит фактора XI |
| Дефицит фактора XII |
| Дефицит фактора XIII |
| Железодефицитная анемия |
| Закономерности опухолевой прогрессии |
| Иммунные гемолитические анемии |
| Клоповое происхождение гемобластозов |
| Лейкопении и агранулоцитозы |
| Лимфосаркомы |
| Лимфоцитома кожи (болезнь Цезари) |
| Лимфоцитома лимфатического узла |
| Лимфоцитома селезенки |
| Лучевая болезнь |
| Маршевая гемоглобинурия |
| Мастоцитоз (тучноклеточный лейкоз) |
| Мегакариобластный лейкоз |
| Механизм угнетения нормального кроветворения при гемобластозах |
| Механическая желтуха |
| Миелоидная саркома (хлорома, гранулоцитарная саркома) |
| Миеломная болезнь |
| Миелофиброз |
| Нарушения коагуляционного гемостаза |
| Наследственная a-fi-липопротеинемия |
| Наследственная копропорфирия |
| Наследственная мегалобластная анемия при синдроме Леш — Найана |
| Наследственные гемолитические анемии, обусловленные нарушением активности ферментов эритроцитов |
| Наследственный дефицит активности лецитин-холестерин-ацилтрансферазы |
| Наследственный дефицит фактора X |
| Наследственный микросфероцитоз |
| Наследственный пиропойкилоцитоз |
| Наследственный стоматоцитоз |
| Наследственный сфероцитоз (болезнь Минковского-Шоффара) |
| Наследственный эллиптоцитоз |
| Наследственный эллиптоцитоз |
| Острая перемежающаяся порфирия |
| Острая постгеморрагическая анемия |
| Острые лимфобластные лейкозы |
| Острый лимфобластный лейкоз |
| Острый лимфобластный лейкоз |
| Острый малопроцентный лейкоз |
| Острый мегакариобластный лейкоз |
| Острый миелоидный лейкоз (острый нелимфобластный лейкоз, острый миелогенный лейкоз) |
| Острый монобластный лейкоз |
| Острый промиелоцитарный лейкоз |
| Острый промиелоцитарный лейкоз |
| Острый эритромиелоз (эритролейкоз, болезнь Ди Гульельмо) |
| Отдельные формы лейкозов |
| Пароксизмалъная холодовая гемоглобинурия |
| Пароксизмальная ночная гемоглобинурия (болезнь Маркьяфавы-Микели) |
| Парциальная красноклеточная аплазия |
| Патологическая анатомия поражения оболочек |
| Плазмоклеточный острый лейкоз |
| Полиорганная недостаточность |
| Поражение нервной системы |
| Порфирии |
| Принципы разделения злокачественных и доброкачественных опухолей системы крови |
| Приобретенные геморрагические коагулопатии |
| Причины гемобластозов |
| Пролимфоцитарный лейкоз |
| Ретикулез (ретикулогистиоцитоз, нелипидный ретикулоэндотелиоз, болезнь Абта-Леттерера-Сиве) |
| Серповидно-клеточная анемия |
| Серповидно-клеточная анемия |
| Синдром Дайемонда — Блекфана |
| Сублейкемический миелоз |
| Т-клеточный лейкоз-лимфома взрослых |
| Талассемия |
| Талассемия |
| Тромбофилий, связанные с дефицитом антитромбина III |
| Тромбоцитопатии |
| Тромбоцитопении |
| Фолиеводефицитная анемия |
| Хроническая лучевая болезнь |
| Хронический лимфолейкоз |
| Хронический лимфолейкоз (хронический лимфоидный лейкоз) |
| Хронический лимфоцитарный лейкоз |
| Хронический мегакариоцитарный лейкоз |
| Хронический миелоидный лейкоз |
| Хронический миелолейкоз |
| Хронический моноцитарный лейкоз |
| Хронический моноцитарный лейкоз |
| Хронический эритромиелоз |
| Цитостатическая болезнь |
| Энтеропатии и кишечный дисбактериоз |
| Эритремия |
| Эритремия (истинная полицитемия, эритроцитоз, болезнь Вакеза) |
| Эритропоэтическая копропорфирия |
| Эритропоэтическая протопорфирия |
| Эритропоэтические уропорфирии |
| Ювенильный миеломоноцитарный лейкоз |
Патологическая анатомия
Смертельные случаи в связи с успешным лечением П. а. встречаются крайне редко. При вскрытии умерших в остром периоде болезни выявляется общее малокровие, желтушность кожи, серозных и слизистых оболочек, жировая дистрофия миокарда, печени, почек. Кровь в сердце и крупных сосудах жидкая, водянистая. Вследствие выраженной гиперплазии кроветворных элементов и исчезновения жира костный мозг плоских костей, а также диафизов и эпифизов трубчатых костей очень сочный, малиново-красного цвета. Характерны атрофические изменения в жел.-киш. тракте.
Сосочки языка, особенно в области корня, атрофичны, сглажены. Слизистая оболочка языка содержит красноватые участки воспаления, располагающиеся чаще по краям и на кончике, иногда афтозные высыпания, трещины (картина так наз. гунтеровского глоссита). Сходные изменения могут наблюдаться в слизистой оболочке десен, щек, мягкого неба, глотки, пищевода. Постоянно выявляется атрофия слизистой оболочки желудка, наиболее выраженная в области дна; складки слизистой оболочки сглажены или не определяются, стенка желудка истончена, в некоторых случаях имеются полипозные разрастания. Атрофические изменения могут наблюдаться также в слизистой оболочке кишечника. Размеры селезенки — в пределах или несколько больше нормы. Печень увеличена незначительно, плотновата. Вследствие отложения гемосидерина ткань селезенки, печени, иногда почек на разрезе имеет ржавый оттенок. Лимф, узлы небольшие, мягкие. В некоторых: случаях макроскопически в спинном мозге выявляются мелкие очаги некроза с размягчением. Иногда находят мелкоточечные кровоизлияния в. серозных и слизистых оболочках, в коже.
Микропрепарат слизистой оболочки желудка при пернициозной анемии: 1 — атрофия желез, 2 — инфильтрация и склероз собственной пластинки слизистой оболочки; окраска гематоксилин-эозином; X 70.
Микроскопически в красном костном мозге отмечается выраженная гиперплазия клеток эритроидного ряда с наличием среди них большого количества мегалобластов — крупных клеток, имеющих нежноячеистую структуру ядра с отчетливо видимыми ядрышками и широкую зону базофильной или полихроматофильной цитоплазмы. Число клеток лейкоцитопоэза несколько снижено. Мегакариоциты содержатся в достаточном или уменьшенном количестве. Наблюдаются выраженные дистрофические изменения и распад клеток, особенно эритроидного ряда, обилие эритро-и сидерофагов. В слизистой оболочке желудка выявляется картина атрофического гастрита, имеется значительное уменьшение количества желез и железистых клеток, особенно обкладочных. Клетки уменьшены в размерах, пикнотичны, уплощены, вследствие чего просвет желез расширен. Возрастает количество слизеобразующих железистых клеток, встречаются участки кишечной метаплазии поверхностного эпителия. Строма слизистой оболочки склерозирована, инфильтрирована лимфоцитами, плазматическими клетками, единичными сегментоядерными нейтрофильными гранулоцитами (рис.). Изменения наиболее выражены в дне желудка, однако интенсивность их может варьировать в различных участках слизистой оболочки. Атрофические изменения не подвергаются обратному развитию при лечении витамином B12 и сохраняются в период ремиссии заболевания.
При биопсии слизистой оболочки тонкой кишки выявляются укорочение кишечных ворсинок, дистрофические изменения железистых клеток со снижением в них фигур митозов в криптах, лимфоидно-плазмоцитарная инфильтрация стромы. После лечения витамином B12 эти изменения могут исчезнуть. Наряду с атрофическими изменениями наблюдается дистрофия нервных волокон языка, нервных клеток подслизистого сплетения (plexus submucosus, s. Meissneri) и мышечно-кишечного сплетения (plexus myen-tericus, s. Auerbachi).
Характерны дистрофические изменения в задних и боковых столбах спинного мозга, преимущественно в шейном отделе, выражающиеся в очаговом набухании с последующим распадом миелиновых нервных волокон. Слияние мелких очагов приводит к образованию больших участков поражения. В некоторых случаях наблюдаются дистрофические изменения черепно-мозговых (черепных, Т.) и периферических нервов. Имеет место гемосидероз костного мозга, селезенки, печени, лимф, узлов, почек, выраженный в различной степени. В селезенке, лимфатических узлах нередко встречаются очаги экстрамедуллярного кроветворения.
Этиология и патогенез
Ведущим фактором в этиологии П. а. является эндогенная недостаточность витамина B12, возникающая вследствие нарушения его всасывания в результате снижения или полного прекращения секреции внутреннего желудочного фактора (гастромукопротеина), необходимого для связывания и последующей абсорбции витамина B12. П. а. может возникнуть в результате поражения жел.-киш. тракта, напр, воспалительным или злокачественным процессом, а также после субтотального или тотального удаления желудка и после обширной резекции части тонкого кишечника. В редких случаях П. а. развивается при нормальной секреции внутреннего желудочного фактора и обусловлена врожденным отсутствием транскобаламина II (белка плазмы крови), с которым витамин B12 связывается и доставляется в печень и красный костный мозг, или отсутствием в кишечнике белкового акцептора витамина B12, необходимого для поступления витамина B12 из кишечника в кровеносное русло. На роль генетического фактора указывают случаи семейных заболеваний, а также возникновение П. а. на почве врожденного нарушения секреции внутреннего желудочного фактора, наследуемого по аутосомно-рецессивному типу. Об участии иммунных механизмов при П. а. свидетельствует наличие в сыворотке крови у большинства больных П. а. и их родственников антител, направленных против париетальных гландулоцитов желудка, а также антител (IgG) против внутреннего желудочного фактора, к-рые определяются как в париетальных гландулоцитах (при гастробиопсии), так и в цитоплазме плазматических клеток красного костного мозга авторадиографией (см.). О существовании аутоиммунных процессов говорят случаи сочетания П. а. с тиреоидитом Хасимото (см. Хасимото болезнь), при этом одновременно присутствуют антитиреоидные антитела и антитела к париетальным гландулоцитам желудка. Аутоиммунный генез П. а. подтверждает успешное применение кортикостероидов. В ряде случаев П. а. возникает без участия специфических антител.
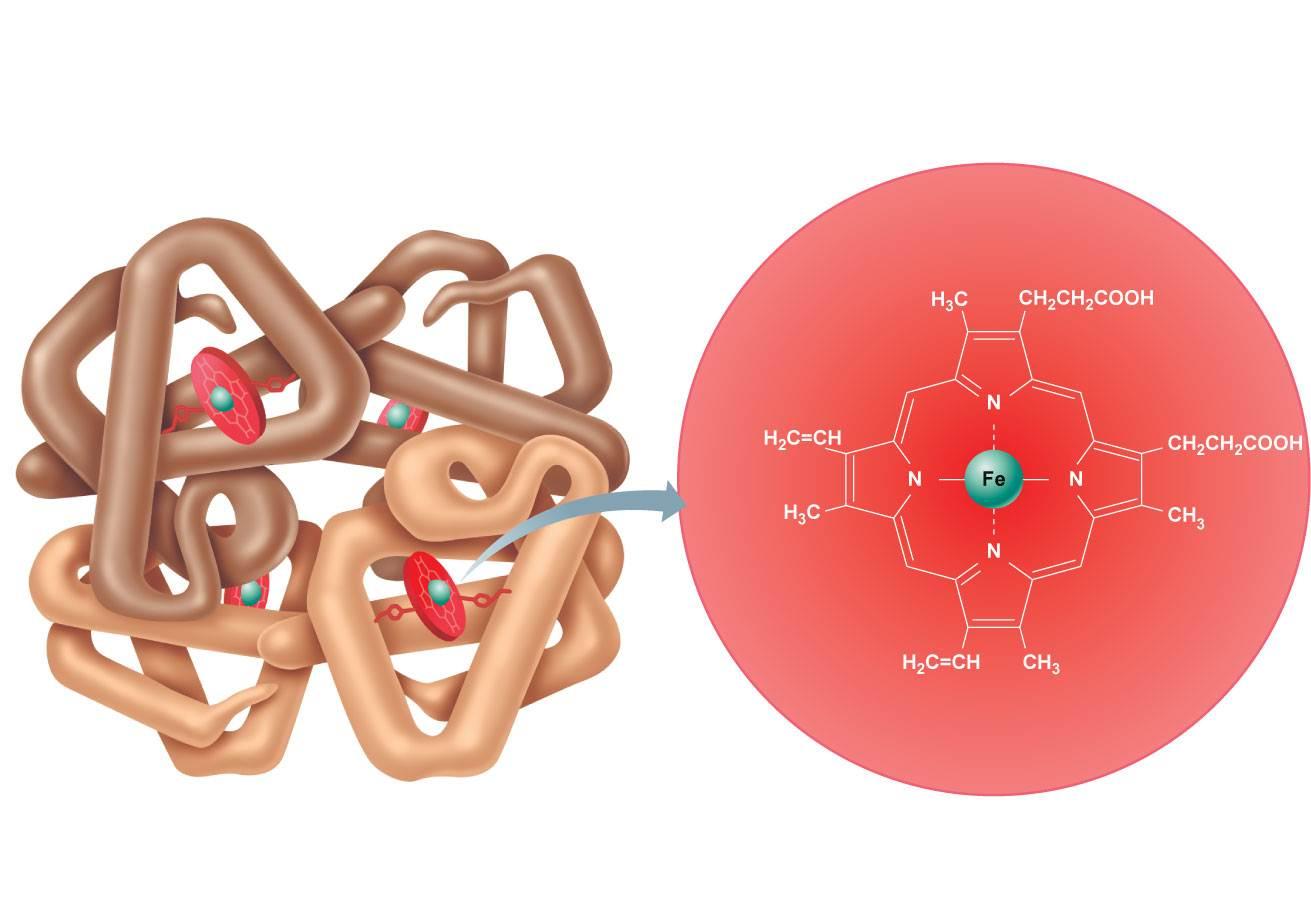
Нарушение кроветворения (см.) характеризуется мегалобластозом красного костного мозга с поражением всех трех ростков кроветворения — эритроидного, гранулоцитарного и мегакариоцитарного. Особенностью мегалобластического эритроцитопоэза является его неэффективность, обусловленная резким нарушением процессов дифференциации эритроидных клеток, в результате которого в красном костном мозге происходит накопление аномальных эритроидных клеток — промегалобластов и мегалобластов. Мегалобластический эритроцитопоэз обусловлен резким снижением активности В ^-зависимых энзимов, участвующих в метаболизме фолатов (солей фолиевой к-ты), необходимых для синтеза ДНК; в частности, снижение активности метилтрансферазы сопровождается кумуляцией в клетках неактивного метилтетрагидрофолата и снижением синтеза ДНК, что приводит к нарушению клеточного деления и развитию мегалобластоза.
Повышенная «интрамедуллярная смертность» мегалобластов, выявляемая при биопсии костного мозга в виде интенсивного эритрофагоцитоза и гемосидероза (см.), ведет к резкому снижению продукции эритроцитов, т. е. к анемии. В то же время в результате повышенного распада гемоглобинизированных мегалобластов (полихроматофильных и оксифильных) возникают симптомы гемолитической анемии (см.) — не-конъюгированная гипербилирубинемия, плейохромия (увеличенное содержание пигментов) желчи, уробилинурия, повышенное выделение стеркобилина с калом. Не исключена возможность участия антител в костномозговом разрушении эрит-рокариоцитов.
Этиология и патогенез
Железодефицитные анемии могут быть обусловлены рядом причин. Повторные кровопотери, даже необильные и скрытые, ведут к потере железа, истощению его резервов в организме и развитию латентного дефицита железа, а затем хронической постгеморрагической Железодефицитной анемии. Причиной кровопотерь часто являются маточные кровотечения, реже жел.-киш., почечные, легочные и др.
Железодефицитная анемия может развиваться под влиянием экзогенных факторов. Так, анемия, связанная с недостатком железа в пище, отмечается у детей, находящихся на однообразном молочном питании, особенно при искусственном вскармливании коровьим или козьим молоком. Для развития Ж. а. у новорожденных может иметь значение дефицит железа у матери во время беременности. В СССР и других экономически развитых странах алиментарная Ж. а. у взрослых встречается редко — при длительной однообразной, преимущественно молочной диете. В развивающихся странах среди необеспеченных слоев населения алиментарная недостаточность может быть причиной анемий гораздо чаще. Другие экзогенные факторы (инфекции, интоксикации, глистные инвазии) влияют на эндогенные механизмы обмена и утилизации железа, что приводит к его перераспределению и развитию относительного дефицита.
Эндогенная недостаточность железа может быть следствием повышенного расхода железа в периоды роста и созревания организма, а также беременности, лактации. Причиной Ж. а. у девушек в пубертатном периоде (ранний, или ювенильный, хлороз) может явиться повышение потребности организма в железе в связи с ростом, появлением менструальных кровотечений, угнетающим действием эстрогенных гормонов на синтез гема. Ж. а. у взрослых женщин обычно связана с маточными кровотечениями (дисфункциональными или на почве фибромиомы матки). Нарушение абсорбции железа и других веществ, важных для кроветворения, происходит также после резекции желудка или части тонкой кишки.
Железодефицитная анемия может быть обусловлена как каждым из перечисленных факторов в отдельности, так и одновременным сочетанием всех или ряда из них. Возможно развитие Ж. а. на фоне хрон, воспалительного процесса, бластоматозов (ранее они объединялись понятием «симптоматические хлоранемии»). В этих случаях, помимо дефицита железа, который носит перераспределительный характер, имеет место миелотоксическое действие основного процесса на гемопоэз.
Патогенез Ж. а. заключается в нарушении синтеза гема из-за недостатка железа в организме и развитии истинного дефицита железа (с истощением его резервов) или в нехватке железа для эритропоэза вследствие перераспределения.
Гейнрих (Н. С. Heinrich, 1970) различает следующие стадии развития дефицита железа: 1) прелатентный дефицит железа, определяемый гистохимически по отсутствию гемосидерина в макрофагах ретикулогистиоцитарной системы костного мозга, а косвенно — на основании повышенной кишечной абсорбции железа; 2) латентный дефицит железа, характеризующийся снижением концентрации сывороточного железа и повышением общей и особенно латентной железосвязывающей способности сыворотки (ОЖСС и ЛЖСС); 3) железодефицитная (гипосидеремическая) анемия с дальнейшим снижением содержания железа в организме и нарушением гемоглобинизации эритроцитов.
Современные представления о патогенезе дефицита железа и развитии Ж. а. опираются на исследования обмена железа в нормальных и патол, условиях. Потеря железа при повторных кровотечениях или недостаточность его всасывания в связи с уменьшением поступления с пищей, а также нарушением абсорбции ведет к уменьшению резервов железа, снижению сывороточного железа и повышению ОЖСС и особенно ЛЖСС с уменьшением процента насыщения переносчика железа— трансферрина (сидерофилина). В результате снижается транспорт железа в костный мозг и уменьшается его включение в клетки эритропоэтического ряда путем микропиноцитоза из ретикулярных клеток или прямой передачи от трансферрина через рецепторные участки ретикулоцитов. При этом снижается поступление в клетку железа, необходимого для синтеза гема.
Следствием нехватки железа для синтеза гема является увеличение протопорфирина в эритроцитах при некоторых формах Ж. а. В условиях дефицита железа резервы его мобилизуются для нужд эритропоэза, что ведет к уменьшению, в частности, костномозгового депо железа. Нарушение образования гема и гемоглобинизации эритробластов является конечной фазой недостаточности эритропоэза, вызванной истинным или перераспределительным дефицитом железа в организме.
Лечение Железодефицитной анемии:
Во всех случаях железодефицитной анемии необходимо установить непосредственную причину возникновения данного состояния и по возможности ликвидировать ее (чаще всего устранить источник кровопотери или провести терапию основного заболевания, осложнившегося сидеропенией).
Лечение железодефицитной анемии должно быть патогенетически обоснованным, комплексным и нацеленным не только на ликвидацию анемии как симптома, но и на ликвидацию дефицита железа и восполнение его запасов в организме.
Программа лечения железодефицитной анемии:
— устранение причины железодефицитной анемии;
— лечебное питание;
— ферротерапия;
— профилактика рецидивов.
Больным железодефицитными анемиями рекомендуется разнообразная диета, включающая мясные продукты (телятина, печень) и продукты растительного происхождения (бобы, сою, петрушку, горох, шпинат, сушеные абрикосы, чернослив, гранаты, изюм, рис, гречневую крупу, хлеб). Однако невозможно добиться противоанемического эффекта только диетой. Если даже больной будет питаться высококалорийными продуктами, содержащими животный белок, соли железа, витамины, микроэлементы,- можно достичь всасывания железа не более 3-5 мг в сутки. Необходимо применение препаратов железа. В настоящее время в распоряжении врача имеется большой арсенал лекарственных препаратов железа, характеризующихся различным составом и свойствами, количеством содержащегося в них железа, наличием дополнительных компонентов, влияющих на фармакокинетику препарата, различных лекарственных форм.
Согласно рекомендациям, разработанным ВОЗ, при назначении препаратов железа предпочтение отдают препаратам, содержащим двухвалентное железо. Суточная доза должна достигать у взрослых 2 мг/кг элементарного железа. Общая длительность лечения не менее трех месяцев (иногда до 4-6 месяцев). Идеальный железосодержащий препарат должен обладать минимальным количеством побочных эффектов, иметь простую схему применения, наилучшее соотношение эффективность/цена, оптимальное содержание железа, желательно наличие факторов, усиливающих всасывание и стимулирующих гемопоэз.
Показания к парентеральному введению препаратов железа возникают при непереносимости всех пероральных препаратов, нарушении всасывания (неспецифический язвенный колит, энтерит), язвенной болезни желудка и двенадцатиперстной кишки в период обострения, при тяжелой анемии и жизненной необходимости быстрого восполнения дефицита железа. Об эффективности препаратов железа судят по изменениям лабораторных показателей в динамике. К 5-7 дню лечения увеличивается количество ретикулоцитов в 1,5-2 раза по сравнению с исходными данными. Начиная с 10-го дня терапии повышается содержание гемоглобина.
Учитывая прооксидантное и лизосомотропное действие препаратов железа, их парентарельное введение можно сочетать с внутривенным капельным введением реополиглюкина (400 мл — один раз в неделю), который позволяет защитить клетку и избежать перегрузки макрофагов железом. Учитывая значительные изменения функционального состояния мембраны эритроцита, активацию перекисного окисления липидов и снижение антиоксидантной защиты эритроцитов при железодефицитной анемии, необходимо в схему лечения вводить антиоксиданты, мембраностабилизаторы, цитопротекторы, антигипоксанты, такие как a-токоферол до 100-150 мг в сутки (либо аскорутин, витамин А, витамин С, липостабил, метионин, милдронат и др.), а также сочетать с витаминами В1, В2, В6, В15, липоевой кислотой. В некоторых случаях целесообразно применение церулоплазмина.
Список препаратов, которые применяют при лечении железодефицитной анемии:
— Жектофер (Jectofer);
— Конферон (Conferon);
— Мальтофер (Maltofer);
— Сорбифер дурулес (Sorbifer durules);
— Тардиферон (Tardiferon);
— Ферамид (Ferramidum);
— Ферро-градумет (Ferro-gradumet);
— Ферроплекс (Ferroplex);
— Ферроцерон (Ferroceronum);
— Феррум лек (Ferrum lek).
— Тотема (tothema)