Синдром прадера-вилли: как распознать патологию развития?
Содержание:
Клиническая характеристика синдрома Прадера-Вилли
Дети с этой наследственной болезнью обычно рождаются доношенными. Часто у них наблюдается нерезко выраженная внутриутробная гипотрофия (низкая подвижность плода). Врачи различают 2 фазы синдрома Прадера-Вилли:
- Первая характеризуется выраженной мышечной гипотонией. Часто ее дополняют: рефлекс Моро, снижение сухожильных рефлексов, тенденция к гипотермии;
- Вторая фаза болезни обычно развивается через несколько месяцев и проявляется в виде полифагии. Дети не могут утолить свой голод, поэтому они готовы есть непрерывно, что приводит к развитию ожирения. При этом жир откладывается чаще всего на проксимальных отделах конечностей и на туловище, в то время как кисти и стопы диспропорционально маленькие. Во второй фазе признаки гипотонии несколько уменьшаются.
У таких детей рост обычно снижен. У мальчиков возникает гипоплазия мошонки, полового члена, крипторхизм. Девочкам характерны гипоплазия половых губ, а в более взрослом возрасте –аменорея и гипоплазия матки.
Как правило, у больных детей психомоторное развитие отстает от нормы. У них хорошая зрительная долговременная память, и они могут научиться читать, но их собственная речь обычно отстает от понимания. Математические навыки, слуховая память и навыки письма чаще всего развиты хуже, что требует особого подхода при обучении. Синдром Прадера-Вилли у детей характеризуется затрудненной речью и маленькимсловарным запасом. Как правило, больные доброжелательны по характеру, плохо умеют управлять своими эмоциями, безынициативны, и им свойственны резкие перепады настроения.
Помимо этих основных симптомов может наблюдаться:
- Высокое арковидное небо;
- Сухая слизистая полости рта;
- Микроцефалия;
- Кариес и дефекты эмали;
- Гипоплазия хрящей ушных раковин;
- Судороги и страбизм;
- Микродонтия;
- Мезобрахифалангия;
- Сколиоз;
- Клинодактилия;
- Синдактилия;
- Нарушение координации;
- Поперечная ладонная складка.
Также нередко в период совершеннолетия синдрому сопутствует сахарный диабет, аномальная гибкость и жидкие лобковые волосы. Обычно у больных встречается не больше пяти указанных признаков.
Общими внешними признаками взрослых с синдромом Прадера-Вилли являются:
- Широкий и большой нос;
- Избыточный вес с жировыми отложениями в центральной части тела;
- Чувствительная кожа, на которой легко образуются синяки и царапины;
- Маленькие ноги и руки с непропорционально узкими пальцами.

Профилактика синдрома
Предотвратить врожденное заболевание невозможно, главное в этом случае – не допустить появления осложнений. Лечение синдрома следует начать как можно раньше, тогда ребенку будет проще приспособиться к обучению в школе и к жизни в обществе.
К профилактике заболевания можно отнести медико-генетические консультации семей, у которых есть предрасположенность к возникновению синдрома. Будущим родителям необходимо провести дородовое генетическое исследование, которое поможет определить особенности строения хромосом плода.
Чтобы улучшить жизнь ребенка с СПВ, следует обеспечить постоянное сотрудничество специалистов медицинских учреждений, родителей и самого малыша.
Эндокринные нарушения
Существует несколько факторов, подтверждающих концепцию дефицита роста у лиц подверженных синдрому.
- Лица, подверженные болезни, имеют низкий рост и страдают от чрезмерного ожирения, то есть у них пониженное содержание свободной жировой массы, пониженная плотность костной ткани, низкий уровень использованной энергии.
- Для данной болезни характерны нарушения в мочеполовой системе. У мужского пола проблемы возникают с неопущением яичек (со временем яички могут опуститься до нормального уровня), а у женского в появлении адренархе. В обоих случаях возможно решение проблемы хирургическим путем.
Нейро-когнитивные отклонения
Синдром Прадера-Вилли создает сложности в процессе учебного воспитания и усвоения материала. Ученые в начале 90-х гг. проводили наблюдения за процессами обучения и трудностями, возникающими при усвоении материала.
У детей, подверженных патологии, особенные когнитивные качества. У них хорошо развито зрительное восприятие, они имеют хороший запас слов и проявляют способности к чтению, но развитие речи значительно уступает визуальному восприятию.
Плохо развито слуховое восприятие и осмысление информации, проблемным становится обучение письму и усвоение математических наук. Нарушена слуховая и зрительная память, низкая концентрация восприятия звуков. В некоторых случаях интеллектуальные способности и возможности со временем улучшаются, но в вышеперечисленных симптомах проблемы будут проявляться.
Синдром Ди Джорджи
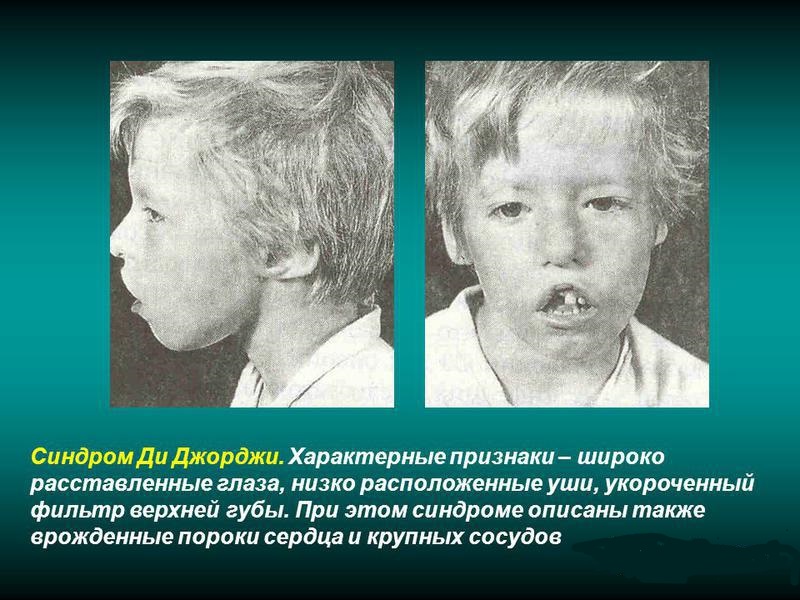
При синдроме Ди Джорджи у больных отмечается наличие врожденной формы аплазии паращитовидных желез и тимуса. Является разновидностью идиопатического изолированного гипопаратиреоза. Встречается достаточно редко.
При этом заболевании патологические изменения касаются околощитовидных (паращитовидных) желез, у которых отмечается дисгенез или агенезия. Вилочковая железа (тимус) отсутствует от рождения. В результате сочетания таких патологий происходит резкое снижение числа Т-лимфоцитов, формируется иммунологическая недостаточность. Кроме того, этот синдром сопровождается формированием врожденных аномалий крупных сосудов.
Заболевание является аутосомным и определяется наличие мутации в 22 хромосоме. В большинстве случаев причиной является спорадическая делеция 22q11 (реже микроделеция 22q11.2). Наследование происходит по доминантному принципу, с полом не связано. Некоторые авторы не соглашаются с такой характеристикой и приводят аргументы в пользу аутосомно-рецессивного типа, обладающего различной эспрессивностью.
Для заболевания характерно нарушение процесса эмбриогенеза 3-4 жаберных карманов, что приводит к нарушению закладки вилочковой железы и паращитовидных желез.
В клинике наиболее постоянными симптомами являются кандидомикоз и гипопаратиреоз, довольно часто сопровождающиеся нарушением процесса формирования рта, носа и ушей.
Тимус из-за нарушения развития в эмбриональном периоде остается неразвитым. Эпителий тимуса не обеспечивает нормального процесса развития Т-клеток. В итоге формируется специфическая форма иммунодефицита, при которой ослабляется гуморальный иммунный ответ и ответ на клеточном уровне. Если у ребенка имеется подобное патологическое нарушение иммунитета, то он будет обладать повышенной чувствительностью к инфекциям бактериального, вирусного и грибкового происхождения.
Синдром может протекать в форме генетически обусловленного отсутствия паращитовидных желез или изолированной недостаточности околощитовидных желез – в сопровождении гипокальциемических судорог, которые начинаются от рождения. Иммунологическая недостаточность приводит к появлению различных инфекционных заболеваний. Как правило, совокупность симптомов вызывает сердечную недостаточность. Кроме того, летальный исход вызывают инфекционные болезни.
Диагностика синдрома предполагает выявление типичных для синдрома патологий: искажения формы лица и черепа, наличие иммунологической недостаточности, аплазии тимуса, дисгенезии или агенезии паращитовидных желез. Ярче всего при заболевании проявляются кандидомикоз и гипопаратиреоз.
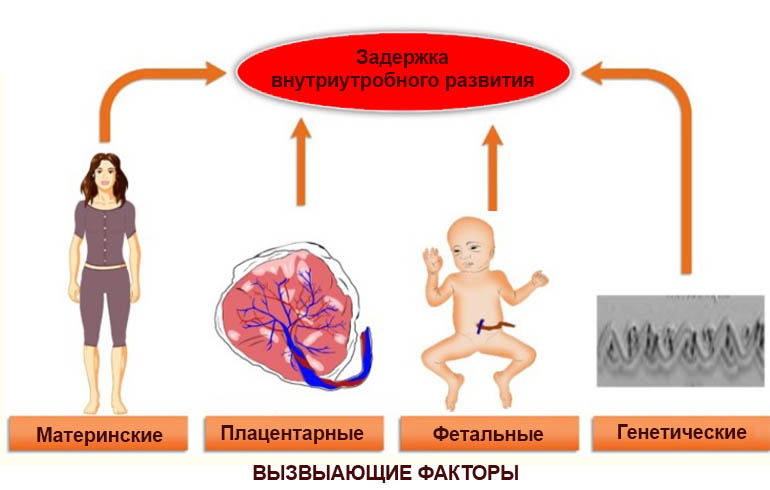
Когда и почему возникают генетические патологии плода: риски по возрастам
Аномалии развития плода закладываются уже в момент оплодотворения сперматозоидом яйцеклетки. Например, такая патология, как триплоидия (наличие трех хромосомом в ряду цепочки, а не двух, как положено), возникает в случае проникновения в яйцеклетку двух сперматозоидов, каждый из которых оставляет по одной хромосоме. Естественно, с таким набором живой организм не может выжить, поэтому на определённом этапе происходит выкидыш или замершая беременность.
В целом хромосомные патологии разделяются на 4 группы:
- Гаметопатия. Патология имеется ещё до зачатия в самом сперматозоиде или яйцеклетке, т.е. это генетическое заболевание — врожденная патология.
- Бластопатия. Аномалии возникают в первую неделю развития зиготы.
- Эмбриопатия. Повреждения эмбрион получает в период от 14 до 75 дней после зачатия.
- Фетопатия. Заключается в формировании патологии развития плода начиная с 75 дня после оплодотворения.
Данные статистики наводит на мрачные мысли. Так, риск рождения малыша с хромосомными аномалиями у 20-летних женщин составляет 1:1667, а у 35-летних уже 1:192. А на деле это означает, что в 99,5% случаев ребёнок у тридцатипятилетней матери родится здоровым.
Что бы вы сказали семьям, которые столкнулись с синдромом?
Марина В.: Когда родители только узнают о диагнозе, они пытаются предугадать будущее: а как будет у нас, пойдём на поправку или состояние ухудшится? Мне очень хочется, чтобы люди не теряли время, приняли ситуацию и проводили время с детьми. Дети быстро растут, та любовь, которая есть здесь и сейчас, должна занимать сто процентов нашего времени и сто процентов нашего внимания.
Марина А.: Есть такая фраза: «То, что вы можете воспринимать спокойно, больше не управляет вами»
Я думаю, она применима в нашей ситуации: важно уделять время своим переживаниям, но не допускать, чтобы они управляли вами. . Я советую изучить информацию, проанализировать её и понять, что это не беда, не ужасная проблема, а обстоятельства
Они так сложились, и мы никак не можем повлиять на этот факт. Но мы можем крепко любить детей, можем прикладывать все усилия, чтобы у них была полноценная жизнь. Я верю, что прогресс малыша, несмотря на исходные данные, на 50% зависит от настроя родителей
Я советую изучить информацию, проанализировать её и понять, что это не беда, не ужасная проблема, а обстоятельства. Они так сложились, и мы никак не можем повлиять на этот факт. Но мы можем крепко любить детей, можем прикладывать все усилия, чтобы у них была полноценная жизнь. Я верю, что прогресс малыша, несмотря на исходные данные, на 50% зависит от настроя родителей.
Общество и культура
1680 Картина Хуан Карреньо де Миранда из Eugenia Мартинеса Вальехо , девушка предположительно иметь PWS
Несмотря на свою редкость, PWS часто упоминается в популярной культуре, отчасти из-за любопытства, связанного с ненасытным аппетитом и увлечением фирменным ожирением, симптомом этого синдрома.
Синдром несколько раз был показан и задокументирован на телевидении. Выдуманный человек с PWS, фигурирующий в эпизоде «Dog Eat Dog» телесериала CSI: Crime Scene Investigation , который транслировался в США 24 ноября 2005 года. В июле 2007 года Channel 4 показал документальный фильм 2006 года под названием Can’t Stop Eating , окружает повседневную жизнь двух людей с PWS, Джо и Тамары. В эпизоде « Extreme Makeover: Home Edition» 2010 года Шерил Кроу помогла Тай Пеннингтону восстановить дом для семьи, младший сын которой, Итан Старквезер, страдал этим синдромом. В эпизоде Mystery Diagnosis на канале Discovery Health 2012 года Конор Хейбах, страдающий синдромом Прадера-Вилли, поделился своим диагнозом.
Причины патологий плода: что влияет на рождение детей с генетическими отклонениями
К фактором, способствующим рождению детей с генетическими аномалиями, относятся:
- Генетическая предрасположенность. Гены — это информация, закладываемая от обоих родителей. Определяются такие показатели, как рост, цвет глаз и волос. Точно также закладываются и различные отклонения, если у обоих или у одного из родителей имеется повреждённый ген. Вот почему запрещается вступать в брак близким родственникам. Ведь тогда возрастает вероятность вынашивания плода с генетической патологией. С партнером, имеющим противоположный генетический набор, больше шансов родить здорового малыша.
- Возраст родителей. К группе риска относятся мамы старше 35 лет и папы старше 40 лет. С возрастом снижается иммунитет, возникают хронические заболевания, и иммунная система женщины попросту “не заметит” генетически повреждённого сперматозоида. Произойдёт зачатие, и, если у молодой женщины организм сам отторгнет неполноценный плод, у возрастной мамы беременность будет проходить более спокойно.
- Вредные привычки мамы. Практически 90% патологических беременностей проходит при маловодии. У курящей женщины плод страдает от гипоксии, продукты распада альдегидов (спиртов) на начальных сроках беременности приводят к мутациям и отклонениям. У алкоголичек в 46% случаев дети рождаются с генетическими патологиями. Спирты также “ломают” генетические цепочки и у отцов, которые любят выпить.
- Инфекции. Особенно опасны такие заболевания, как грипп, краснуха, ветрянка. Наиболее уязвимым плод является до 18-й недели, пока не сформируется околоплодный пузырь. В некоторых случаях женщине предлагают сделать аборт.
- Приём медикаментов. Даже обычный ромашковый чай для беременной женщины является токсичным. Любой приём лекарств должен сопровождаться консультацией врача.
- Эмоциональные потрясения. Они вызывают гибель нервных клеток, что неизменно сказывается на развитии плода.
- Плохая экология и смена климата. Забеременев во время отдыха на Таиланде, есть вероятность вместе с беременностью привезти опасную инфекцию, которая в родных краях начнет медленно развиваться, сказываясь на здоровье малыша.
Хадассе нет равных в диагностике орфанных заболеваний
Для обнаружения орфанных заболеваний в медицинском центре используются самые современные генетические тесты, опираясь на которые специалисты иерусалимской больницы безошибочно обнаруживают любые известные генные мутации, при своевременном обращении – на самой ранней стадии. Но это еще не все: врачам Хадассы также нередко удается пополнить длинный список редких болезней.
Для получения более точной информации о стоимости лечения и специальных предложениях нажмите кнопку
Так в апреле 2017 года в одном из самых престижных международных изданий в области генетики American Journal of Human Genetics появилась публикация о результатах исследований, проведенных группой ученых под руководством заведующей отделением генетических и метаболических заболеваний МЦ Хадасса профессора Орли Эльпелег. Исследовательская группа сумела впервые описать неизвестное до сих пор опасное генетическое заболевание, поражающее нервную систему ребенка, и разработать методы его раннего обнаружения.
Профессор Орли Эльпелег
Эксперт мирового уровня в области клинической генетики, педиатр, заведующая отделением генетических и метаболических заболеваний МЦ Хадасса.
Благодаря безошибочным диагнозам профессора Эльпелег были спасены сотни детей.
Читать резюме врача Записаться на консультацию
А как складываются отношения Роберта и Кристины с другими вашими детьми?
Марина А.: Старший сын, Леон, ждал братика. Целовал живот, когда я была беременна. Он с такой нежностью к нему относится с самого рождения. Во время кормлений, когда я через трубку заливала Роберту молочко, Леон сидел рядом и держал братика за руку, очень ответственно подходил к этому процессу. А ведь у них разница в возрасте меньше двух лет. И сейчас он так радуется успехам брата. Например, когда Роб пытается стоять, Леон прибегает и рассказывает мне: «Ты представляешь, Роберт стоял! Я считал до трёх, а он стоял!» Я очень рада, что у Роберта есть старший брат, он огромная поддержка для всех.
 Леон с младшим братом Робертом
Леон с младшим братом Робертом
Марина В.: Кристина — мой пятый ребёнок, и другие дети видят, что она отличается, задают вопросы. Я считаю, что главное в отношении с детьми — честность, поэтому я им всё объясняю. Они мне помогают следить за ней, играют с ней, делают вместе развивающие зарядки. Волнуются за неё, когда ей нужно уколы ставить. Мы их называем уколами силы. Вся семья вовлечена.
Лечение синдрома Прадера-Вилли
Для нормализации строения тела и обменных процессов назначают:
- строгую низкокалорийную диету. Запрещенные продукты должны полностью отсутствовать в семье. К ним относятся: все сладости, мучное, жирные и жареные блюда, соусы, макароны, белый рис, картофель, манка, сладкие напитки, виноград, сухофрукты, шоколад;
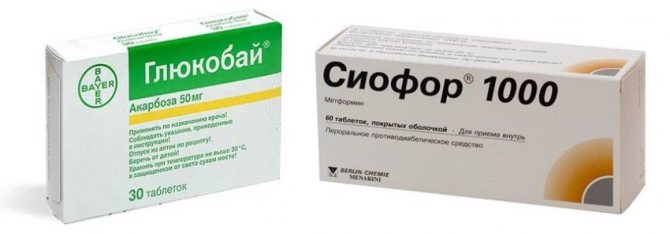
- препараты для снижения сахара в крови: «Сиофор», «Глюкобай»;
- стимулятор образования гонадотропинов «Клостилбегит»;
- гормон роста оптимально начать вводить до 2-х лет. Помимо непосредственного увеличения длины тела улучшается двигательная активность, структура костей, тормозится ожирение. Рекомендуют низкие дозы с постепенным увеличением под контролем ИРФ в крови.
К общеукрепляющему лечению относятся:
- общий массаж;
- рефлексотерапия;
- лечебная физкультура;
- душ Шарко, подводный душ-массаж;
- применение витаминных комплексов, коэнзима Q10 («Убихинон»), нейротропных средств («Пантогам», «Энцефабол»).
К обучению детей нужен особый подход, они могут понимать и усваивать материал достаточно хорошо, но затрудняются в высказывании и написании своих знаний.
Уход
PWS неизлечим; Доступны несколько методов лечения для уменьшения симптомов заболевания. В младенчестве субъекты должны проходить курс лечения для улучшения мышечной силы. Также показаны логопедия и трудотерапия. В школьные годы дети получают пользу от хорошо структурированной учебной среды и дополнительной помощи. Самая большая проблема, связанная с синдромом, — сильное ожирение. Доступ к продуктам питания должен строго контролироваться и ограничиваться, обычно путем установки замков на всех местах хранения продуктов, включая холодильники. Физическая активность людей с СПВ любого возраста необходима для повышения силы и пропаганды здорового образа жизни.
Назначение ежедневных инъекций рекомбинантного GH показано детям с PWS. GH поддерживает линейный рост и увеличение мышечной массы, а также может уменьшить озабоченность едой и прибавку в весе.
Из-за тяжелого ожирения обструктивное апноэ во сне является частым осложнением , и часто требуется установка положительного давления в дыхательных путях . Человеку, у которого был диагностирован СПВ, возможно, придется пройти хирургическое вмешательство. Одна операция, которая оказалась безуспешной для лечения ожирения, — это желудочный анастомоз.
Поведенческие и психические проблемы должны выявляться на ранней стадии для достижения наилучших результатов. Эти проблемы лучше всего решать с помощью родительского образования и обучения. Иногда также вводятся лекарства. Агонисты серотонина оказались наиболее эффективными в уменьшении приступов гнева и улучшении компульсивности.
Общие сведения о синдроме Вилли-Прадера
Первое упоминание о патологии датируется 1887 годом. Лэнгдон Даун описал девочку-подростка, у которой отмечалась задержка физического развития, гипогонадизм и ожирение. Первоначально болезнь получила название «полисарция». Полноценную характеристику синдрому дали швейцарские врачи Прадер, Вилли и Лабхарт в 1956 году. Позднее в ходе глубокого изучения, доктора определили и точную локализацию генетической мутации, которая приводила к возникновению заболевания у детей. Они также связали изменения с синдромом Ангельмана. Оба расстройства провоцируются дефектом в строении 15 хромосомы. При этом в одном случае аномалия формируется в материнской копии, а в другом – в отцовской. Патология была названа синдромом Вилли-Прадера в честь врачей, внесших самый большой вклад в ее изучение. Болезнь относится к числу редких, так как ее распространенность колеблется в пределах одного случая на 10–25 тысяч новорожденных. Половой или расовой предрасположенности не установлено.

Диагностика
Он традиционно характеризуется гипотонией, низким ростом, гиперфагией, ожирением, поведенческими проблемами (особенно поведением, подобным обсессивно-компульсивному расстройству ), маленькими руками и ногами, гипогонадизмом и легкой умственной отсталостью. Однако при ранней диагностике и раннем лечении (например, с помощью терапии гормоном роста) прогноз для людей с СПВ начинает меняться. Как и аутизм, СПВ представляет собой расстройство спектра, симптомы которого могут варьироваться от легких до тяжелых и могут меняться на протяжении всей жизни человека. Поражаются различные системы органов.
Традиционно СПВ диагностировали на основании клинических проявлений. В настоящее время синдром диагностируется с помощью генетического тестирования; Рекомендуется тестирование новорожденным с выраженной гипотонией. Ранняя диагностика СПВ позволяет своевременно вмешаться и назначить гормон роста на ранней стадии . Детям с СПВ показаны ежедневные инъекции рекомбинантного гормона роста (ГР). GH поддерживает линейный рост и увеличение мышечной массы, а также может уменьшить озабоченность едой и прибавку в весе.
Основой диагностики является генетическое тестирование , в частности, тестирование метилирования на основе ДНК для выявления отсутствия отцовской области PWS / AS на хромосоме 15q11-q13. Такое тестирование выявляет более 97% случаев
Тестирование, специфичное для метилирования, важно для подтверждения диагноза СПВ у всех людей, но особенно у тех, кто слишком молод, чтобы проявлять признаки, достаточные для постановки диагноза на клинических основаниях, или у тех людей, у которых есть атипичные результаты.
PWS часто ошибочно диагностируется как другие синдромы из-за того, что многие в медицинском сообществе не знакомы с ним. Иногда его ошибочно принимают за синдром Дауна просто из-за относительной частоты синдрома Дауна по сравнению с PWS.
Уникальные специалисты по ТКМ
В некоторых случаях ТКМ является единственным методом эффективного лечения орфанных заболеваний. В МЦ Хадасса эту процедуру проводят всемирно известные специалисты, не имеющие конкурентов в Израиле.
Профессор Полина Степенски
Онкогематолог со стажем более 19 лет, специалистка мирового уровня в области трансплантации костного мозга.
Профессор Степенски провела около 500 успешных ТКМ.
Читать резюме врача Записаться на консультацию
Доктор Ирина Зайдман
Педиатр и онкогематолог со стажем более 17 лет, заведующая педиатрическим отделением ТКМ клиники Хадасса.
Работая в Израиле и за рубежом, провела более 400 успешных ТКМ.
Читать резюме врача Записаться на консультацию
Синдром Прадера-Вилли
Синдром Прадера-Вилли- врожденное заболевание, при котором возникает сочетание ожирения, низкого роста, снижения функции половых желез (гипогонадизм) и низкого интеллекта. Это заболевание имеет очень широкий спектр проявлений и признаков. Течение болезни отличается в каждом отдельном случае и может варьировать от легкой формы до тяжелой, которая прогрессирует в течение всей жизни человека.
Заболевание впервые описано швейцарскими педиатрами А. Prader и H. Willi в 1956 г. и встречается у 1 человека на 25000-10000 новорожденных. Причиной данного генетического заболевания является отсутствие или недостаточное функционирование некоторых генов (или их частей) на 15 отцовской хромосоме. Следует отметить, что с помощью обычного исследования хромосомного состава кариотипа выявить данную патологию невозможно. Для этого применяются специальные цитогенетические и молекулярно-генетические методы.
Дети с синдромом Прадера — Вилли обычно рождаются доношенными с незначительной внутриутробной гипотрофией и нередко в асфиксии. В 10-40% случаев наблюдается ягодичное предлежание. Заболевание характеризуется выраженной мышечной гипотонией при рождении, сохраняющейся в течение первого года жизни ребенка. Сосательный и глотательный рефлексы снижены, что затрудняет кормление ребенка. Из-за гипотонии у таких детей задерживается развитие двигательных функций: они с трудом учатся держать голову, сидеть и т. д. Мышечная гипотония постепенно уменьшается и к школьному возрасту почти полностью исчезает.
Позднее, к второму-четвертому году жизни появляются постоянное чувство голода и отсутствие насыщения, приводящие к развитию ожирения, причем отложение жира наблюдается преимущественно на туловище и в проксимальных отделах конечностей. Из-за тяжелого ожирения грозным осложнением является обструктивное апноэ (остановка дыхания) во сне.
Рост больных нередко снижен. Часто отмечается долихоцефалия (удлиненная форма головы), миндалевидный разрез глаз, низко расположенные ушные раковины, широкая переносица, маленький рот с тонкой верхней губой. Стопы и кисти больных диспропорционально маленькие (акромикрия). У 75% детей наблюдается слабая пигментация кожи, волос и радужки.
У мальчиков при рождении отмечается недоразвитие полового члена, мошонки,крипторхизм, а у девочек недоразвитие половых губ, иногда и матки. В дальнейшем заболевание проявляется задержкой или отсутствием полового созревания, бесплодием.
Психомоторное развитие отстает от возрастной нормы — коэффициент интеллектуального развития — от 20 до 80 ед. (при норме 85-115 ед.). Как правило, дети с синдромом Прадера-Вилли имеют хорошую долговременную зрительную память, они могут научиться читать, могут обладать богатым пассивным словарем, но их собственная речь обычно хуже, чем понимание. Слуховая память, математические навыки и навыки письма, зрительная и слуховая кратковременная память у таких детей обычно значительно хуже. Больные доброжелательны, настроение характеризуется частой сменой. Описаны нарушения координации, судороги, косоглазие. Продолжительность жизни больных может достигать 60 лет и более. Нередко у таких детей развивается сахарный диабет.
Лечение Синдром Прадера-Вилли является врожденной генетической аномалией и, следовательно, не может быть излечен. Однако если диагностировать данное заболевание на раннем этапе и начать его лечение, то прогноз развития заболевания становится более оптимистичным.
Младенцы со сниженным мышечным тонусом должны получать массаж и другие виды специальной терапии. Комплекс лечебных мероприятий включает также диету с ограничением жиров и углеводов и препараты, способствующие формированию вторичных половых признаков (гонадотропины). Рекомендуется терапия гормоном роста.
Рекомендуется использование специальных методик развития ребенка, занятия с логопедом и дефектологом.
Медико-генетическое консультирование Родителям ребенка с синдромом Прадера-Вилли рекомендуется пройти генетическое обследование, прежде чем планировать дальнейшую беременность, поскольку существует риск того, что следующий ребенок у тех же родителей родится также с синдромом Прадера-Вилли, что зависит от механизма, вызвавшего генетический сбой.