Половые хромосомы
Содержание:
Половые хромосомы у цветковых растений
Для большинства цветущих растений или покрытосеменных растений мужские и женские половые органы присутствуют на одном и том же цветке. Иногда один растение может производить отдельные мужские и женские цветы для усиления креста оплодотворение или мужские и женские половые органы могут созревать в разное время. Тем не менее, присутствие различных мужских и женских растений встречается относительно редко, и только шесть процентов покрытосеменных растений показывают эту характеристику, которая называется диоэксизмом. Даже те, у кого этот вид полового диморфизма возрастает из-за стерильных мужских или женских стерильных мутаций и, следовательно, различных половых хромосом, известны только в четырех семействах растений.
Похоже, что растения находятся на ранних стадиях эволюции гетероморфных половых хромосом. Поэтому они могут быть использованы в качестве моделей для изучения событий, которые приводят к определению хромосомного пола.
У-хромосома и наследственные заболевания
Как отмечалось ранее, мужская хромосома отличается от Х-хромосомы как размерами (она меньше), так и формой (имеет вид крючка). Также для нее специфичен и набор генов. Так, мутация одного из генов У-хромосомы фенотипически проявляется появлением пучка жестких волос на мочке уха. Этот признак характерен только для мужчин. Известно такое наследственное заболевание, вызванное хромосомной мутацией, как синдром Клайнфельтера. Больной мужчина имеет в кариотипе лишние женские или мужские хромосомы: ХХУ или ХХУУ.

Подводя итог, отметим, что у человека, как и у других млекопитающих, пол будущего организма определяется в момент оплодотворения, вследствие определенной комбинации в зиготе половых Х- и У-хромосом.
47 хромосом. Диагностика
47 хромосом у человека диагностируется на всех этапах, начиная с беременности. Уже в 1 и 2 триместре можно определить признаки отклонения – для этого существует множество анализов, которые для большей достоверности рекомендуется проводить в комплексе. Кроме того, установить диагноз даунизма у рожденного ребенка можно на основе внешнего осмотра или исследований генетического материала.
Как определить у новорожденных
Диагноз можно сделать с помощью осмотра на характерные внешние признаки, не прибегая к генетическому исследованию. Кроме того, больного синдромом проверяют на наличие сопутствующих заболеваний, которые могут быть типичны для детей с даунизмом. Для более точного результата проводят детальное изучение хромосомного набора, которое заключается в окрашивании хромосом и определении лишних фрагментов.
Как определяют синдром Дауна при беременности
Для диагностики возможного даунизма ребенка во время беременности проводят множество исследований, которые покажут риск рождения больного. Вам останется лишь принять решение, делать прерывание или рожать ребенка с заболеванием. К таким исследованиям относятся:
- Скрининг на генетические аномалии – сочетание биохимического анализа крови и УЗИ.
- УЗИ. Проводится на 11-13 недели, выявляет толщину воротникового пространства и контуры лица, которые у детей-даунов отличаются от здоровых.
- Биохимический анализ крови помогает выявить содержание особых веществ в организме ребенка.
- Амниоцентез – пункция амниотической оболочки для получения образца околоплодных вод.
- Кордоцентез – получение кордового плода для дальнейшего исследования.
- Биопсия ворсин хориона – тест на отклонения развития плода.
Ретинобластома
Ретинобластомой называют злокачественную опухоль сетчатки глаза. Процесс развития начинается обычно в детском возрасте, причем исходным материалом являются ткани эмбрионального происхождения. Пиковая фаза приходится на двухлетний возраст.
Практически все известные случаи выявляются в течение первых 5 лет жизни.
Причиной заболевания в большинстве случаев является мутация в генетическом материале. При этом необходимо наличие генетической обусловленности за счет наличия мутантной версии гена Rb, полученной по наследству. Вторая мутация, вызывающая появление опухоли, происходит в ретинобласте.
Существует небольшая вероятность, что у родителей, которые переболели ретинобластомой, могут родиться дети с отсутствием патологического изменения.
Отмечаются односторонние и двусторонние случаи ретинобластомы. По статистике для двусторонней формы вероятность наследственного происхождения заметно выше.
Симптомы заболевания включают боль в глазах, свечение зрачка, а также потерю зрения. Выявить их у маленького ребенка очень и очень трудно.
Диагностика обычно проходит в форме обследования под наркозом с применением УЗИ, КТ и МРТ. Достаточно распространенным приемом является биопсия красного костного мозга и спинномозговая пункция. По тяжести симптомов выделяется 5 групп.
Существует два эффективных метода лечения. При криотерапии и фотокоагуляции остается возможность сохранить и зрение, и сам глаз. Осложнения при их использовании возникают редко. Тем не менее, если возникнет рецидив, лечение потребуется повторить в той же форме. Обычно криотерапия используется в случаях, когда поврежден передний отдел сетчатки. Для заднего отдела более предпочтительным вариантом представляется фотокоагуляция.
Синдром Шерешевского-Тернера
Является следствием моносомии по хромосоме Х. Больные дети часто рождаются недоношенными или имеют сниженную массу тела. Одним из классических признаков синдрома Шерешевского-Тернера, который можно заметить сразу после рождения, является выраженная кожная складка на шее. Среди других клинических проявлений отмечаются:
- пороки сердца;
- отечность верхних и нижних конечностей;
- нарушение циркуляции лимфы;
- задержка речевого и физического развития.
По мере взросления ребенка проявляются характерные черты строения тела. Рост обычно не превышает 150 см, крыловидные складки на шее сохраняются, ушные раковины могут деформироваться, верхняя челюсть недоразвита, грудная клетка широкая. Моносомия по хромосоме Х влияет на развитие органов половой системы. У женщин отмечается отсутствие фолликулов в яичниках, нарушение менструального цикла, недоразвитие молочных желез. У мужчин снижается уровень тестостерона, может отсутствовать одно или оба яичка либо отмечаться их недоразвитие.
Прогноз при синдроме Шерешевского-Тернера относительно благоприятный. При отсутствии тяжелых пороков развития и регулярном наблюдении у специалиста продолжительность жизни не сокращается.
Лечение ЗПРР при хромосомных заболеваниях.
Основой лечения является уникальная методика патогенетической терапии речевых расстройств при хромосомной патологии — биофизическая активация нейромоторных структур, основу которого составляет щадящая стимуляция проводников нервной системы микротоками с использованием нейрофизиологического прибора. Метод лечения базируется как на активации самих речевых центров, так и на восстановлении нарушенных связей между центрами и полушариями головного мозга. Помимо этого, восстанавливаются разрозненные связи речевых центров с другими областями мозга, участвующими в реализации речевой функции. В процессе лечения формируется физиологичное, последовательное взаимодействие всех зон мозга, связанных с речепродукцией. В результате появляется речь.
Проведение биофизической активации сочетается с дополнительными методиками лечения, такими как — лимфомежклеточная терапия, которая применяется для регулирования интегративной деятельности и восполнения дефицита энергетической системы мозга и позволяющая применять малые дозы церебропротекторов, которые вводятся эндолимфатически и попадают в ткани головного мозга, минуя гематоэнцефалический барьер.
В качестве другого способа использования препаратов с нейротрофическим и антиоксидантным действием применяется методика эндоназального электрофореза кортексина, что позволяет вводить лекарственные препараты непосредственно в ткани головного мозга.
Исследования последних десятилетий выявили, что у большинства детей с речевыми и поведенческими проблемами в различной степени нарушены функции мозжечка и базальных ганглиев. Именно функционирование мозжечка определяет успешность ребенка в обучении. С этой целью применяется уникальная разработка Центра авиакосмической медицины — подошвенный имитатор опорной нагрузки «Корвит», применяемый для нейрофизиологической регуляции стато-кинетической функции ЦНС. В основе терапевтического воздействия аппарата «Корвит» лежит процесс активации опорной афферентации, отвечающей за нормализацию процессов возбуждения и торможения в центральной нервной системе, что приводит к уменьшению спастичности мышц, развитию и закреплению функциональных связей в головном мозге, способствующих восстановлению координации движений, и, опосредованно, улучшению речи и мышления.
Также для успешного лечения различных форм ЗПРР специалистами применяется одно из достижений современной науки — метод аудиовокальной терапии RUSTOMATIS. Прибор использует звукозаписи высокочастотных и низкочастотных компонентов. При чередовании такой музыки путем напряжения и расслабления у ребенка тренируется аппарат среднего уха – молоточек и стремечко, с помощью чего расширяется диапазон восприятия внешних факторов, увеличивается концентрация внимания, в мозг поступает новая информация и, как следствие исчезают многие нарушения и расстройства.
Обязательным звеном в лечебном комплексе у детей с наличием речевых расстройств является занятия с клиническим психологом, а также логопедическая коррекция, которая включает диагностику степени нарушений, ежедневные занятия, направленные на улучшение речевой функции и логопедический массаж для коррекции различных видов дизартрии и дисфагии.
На фоне сочетания проведения биофизической активации со вспомогательными методиками лечения наблюдаются положительные изменения, которые могут быть видны уже через несколько процедур, но максимальный эффект развивается через полтора-три месяца после курса. Как правило, для закрепления полученных результатов и дальнейшего развития двигательных и когнитивных навыков специалистами центра рекомендуется повторный курс лечения через 5-6 месяцев.
Описание
Молекулярно-цитогенетическая диагностика распространённых хромосомных нарушений (анеуплоидий) по 13, 16, 18, 21, 22, X, Y-хромосоме — молекулярно‐цитогенетический метод, позволяющий определять изменения хромосом анеуплоидий (число хромосом, не кратное гаплоидному набору).
Половые хромосомы — X и Y, и аутосомы — 13, 16, 18, 21, 22 — наиболее часты и имеют наиболее негативное влияние на пренатальное развитие человека. Молекулярно-цитогенетическая диагностика
Молекулярно-цитогенетическая диагностика позволяет выявить отклонение в количестве хромосом (анеуплоидия). В эмбрионах с анеуплоидией отсутствует одна хромосома (моносомия), или имеется лишняя хромосома (трисомия). Наиболее распространённой хромосомной патологией является трисомия (три хромосомы вместо двух в норме) по 21-й хромосоме, или синдром Дауна. Возможно рождение живых детей с трисомиями по хромосоме 13 (синдром Патау), хромосоме 18 (синдром Эдвардса). Эти хромосомные болезни характеризуются еще более тяжёлыми, чем при синдроме Дауна, пороками умственного и физического развития
Как правило, такие дети не живут более года.Спонтанное прерывание беременности
Нарушения репродуктивной функции женского организма, которая сопровождается неожиданным прерыванием беременности, могут быть обусловлены действием различных факторов, среди которых важное место занимают генетические. Во многих случаях беременность прерывается из-за хромосомных нарушений при образовании эмбриона, несовместимых с его жизнью
Чаще возникают ситуации, когда в результате нарушения овогенеза (созревания яйцеклетки) или сперматогенеза (формирования зрелых сперматозоидов) возникают клетки с утраченной или лишней хромосомой или образуется зигота (зародыш) с набором хромосом, по количеству превышающим норму (в норме зигота содержит 46 хромосом). Причинами этому могут быть хронический стресс, курение, употребление наркотиков, алкоголя, экологически неблагоприятные ситуации, а также внутренние факторы: наличие в организме матери или отца генов — небольших участков хромосом, предрасполагающих к нерасхождению хромосом в процессе формирования половых клеток. А когда формируется генетически неполноценный зародыш, по законам естественного отбора организм матери пытается избавиться от него.
С помощью молекулярно-цитогенетической диагностики распространённых хромосомных нарушений, исследуя хорион, определяют отклонения в хромосомном наборе. Данные исследования могут быть выполнены, в том числе и новорождённым детям, в этом случае исследуется кровь плода.
На более позднем сроке прерывания беременности проводится исследование наиболее часто встречающихся анеуплодий (изменений числа хромосом) при неразвивающейся беременности по 13, 16, 18, 21, 22, X, Y-хромосомам (7 хромосом).Показания:
- На более поздних сроках прерывания беременности.
- Беременные от 35 лет и старше.
- Наличие в семье ребёнка с хромосомными заболеваниями или врождёнными пороками развития.
- Замершая беременность с установленными хромосомными дефектами у плода.
- В семье есть ребёнок с хромосомной патологией; носителем хромосомной патологии является один из членов семьи.
- У ближайших родственников есть моногенные заболевания (например, муковисцидоз).
- Женщины, перед зачатием или на раннем сроке беременности принимали сильнодействующие препараты (например, противоопухолевые) или перенесли такие инфекции, как гепатит, краснуха, токсоплазмоз.
- Членам семей с отягощённым акушерским анамнезом (невынашивание, самопроизвольные аборты, мертворождение, рождение ребёнка с врождёнными пороками).
- Высокий риск рождения ребенка с хромосомной патологией, рассчитанный по результатам комбинированного скрининга (УЗИ+биохимические тесты).
- Пороки или отклонения развития, выявленные при ультразвуковом исследовании (ультразвуковые маркёры хромосомной патологии).
Подготовка
Условия подготовки к анализу определяются лечащим врачом.
Аномалии AZF-локуса
На Y-хромосоме также имеется особый участок, который контролирует процесс выработки сперматозоидов. Именно от этого зависит, насколько эффективным будет сперматогенез. Кроме того, состояние этого участка сказывается на свойствах сперматозоидов, таких как общее количество в эякуляте, способность двигаться, наличие структурных изменений и способность к оплодотворению. Только при наличии хорошо сформированных подвижных сперматозоидов мужской генетический материал может быть доставлен до яйцеклетки. Иными словами, о состоянии этого небольшого участка генетического кода зависит способность мужчины иметь детей.
При наличии в AZF-локусе аномалий процесс выработки сперматозоидов нарушается. В результате могут развиться азооспермия и олигозооспермия. При этих патологиях в эякуляте либо совсем не содержится сперматозоидов, либо их число сильно снижено.
Сам AZF локус делится на три части со специфическими задачами. Они именуются путем добавления индекса: AZFa, AZFb и AZFc. Возникшая делеция может удалять фрагмент отдельной части, либо ее целиком, либо захватывать сразу два региона. При полном удалении AZF развивается тяжелое поражение сперматогенеза. Частичные делеции могут проявляться по-разному. При этом на степень проявления патологии влияют размеры утраченного фрагмента и его расположение в локусе
Поэтому для прогностических целей крайне важно знать, в каком месте произошла делеция. Кроме того, эта информация может использоваться для правильного планирования семьи и проведения экстракорпорального оплодотворения
Если при делеции был удален весь локус или любой из регионов с индексами a/b, то у мужчины не могут быть получены жизнеспособные сперматозоиды. Если делецию можно описать формулой AZFb/AZFb+, то развивается азооспермия из-за тяжелых нарушений процесса формирования сперматозоидов.
Делеции участка AZFc приводят к проявлению патологических симптомов различной степени тяжести. В том числе возможно развитие олигоспермии, которая в принципе допускает зачатие. В 50-70 процентах от общего числа подобных случаев возможно получение сперматозоидов для дальнейшего использования в методах искусственного оплодотворения. Частичная делеция региона AZFс может выражаться в форме различных нарушений от нормозооспермии до азооспермии.
Все делеции в AZF-локусе, вызывающие ту или иную патологическую ситуацию, являются причинами мужского бесплодия. Определение мутации возможно путем гистологического анализа семенной жидкости. При этом необходима остановка созревания сперматозоидов или обнаружение незрелых сперматозоидов. Для получения точных данных о делециях в AZF-локусе используется ПЦР 6 маркеров, которые относятся к отдельным участкам локуса.
СИНДРОМ КЛЯЙНФЕЛЬТЕРА
- Кариотип 47,ХХY, тест на половой хроматин – положительный. Y-хроматин –положительный.
- Частота встречаемости 1:500 – 1:700 новорожденных мальчиков.
Фенотип мужской, возможна гинекомастия, евнухоидный тип телосложения (ширина таза обычно больше ширины плеч, удлиненные нижние конечности, высокий рост, скудное оволосение), обычно микроорхидизм при нормальном размере полового члена. Возможен крипторхизм. В редких случаях (при увеличении количества Х-хромосом: 48,ХХХY и 49,ХХХХY) – нарушения интеллекта, функции внутренних органов.
В 5-10% случаев наблюдается мозаичная форма (46,ХY/47,ХХY или 46,ХХ/47,ХХY). В редких случаях у таких больных возможно обнаружить сперматогенез (если клетки сперматогенеза относятся к клону с нормальным кариотипом).
Практически все взрослые больные с синдромом Кляйнфельтера бесплодны вследствие азооспермии.
Учитывая, что большинство людей с синдромом Кляйнфельтера имеют мужской фенотип, нормальный интеллект, достаточный уровень потенции, они вступают в брак. Таким образом, показанием для исследования на половой хроматин является олигоазооспермия у пациента.
Позитивное влияние некоторых делеций на жизнеспособность
Небольшие изменения делеционного характера могут существенно повлиять на выживание организма. К примеру, утрата гена, кодирующего белок CCR5-δ32, становится причиной невосприимчивости к вирусу иммунодефицита человека. Ученые предполагают, что эта мутация впервые появилась около 2,5 тысячелетий назад и с течением времени распространилась по территории Европы.
Имеющееся на сегодняшний день распределение отличается неравномерностью. Согласно статистическим данным, около 10% жителей европейских стран устойчиво к ВИЧ. Вместе с тем в скандинавских государствах этот показатель достигает 14-15 процентов. Русские и финны демонстрируют 16-процентный уровень устойчивости. В то же время для Сардинии частота равняется скромным 4 процентам.
Ряд ученых выдвинул гипотезу, что подобное распространение определяется прошедшими в средневековый период эпидемиями бубонной чумы. Вероятно, мутация в гене вызывает повышенную сопротивляемость этому заболеванию. Поэтому на территории тех стран, где прошлась «черная смерть», выжило больше людей с этим генотипом.
Генетическая информация в У-хромосоме
Исследованиями ученых-генетиков, в частности Т-Х. Моргана, было установлено, что у человека и млекопитающих генный состав Х- и У-хромосом неодинаков. Мужские хромосомы у человека не имеют некоторых аллелей, присутствующих в Х-хромосоме. Однако в их генофонде представлен ген SRY, контролирующий сперматогенез, приводящий к формированию мужского пола. Наследственные нарушения этого гена в эмбрионе приводит к развитию генетического заболевания – синдрома Суайра. В результате женская особь, развивающаяся из такого эмбриона, содержит в кариотипе ХУ половую пару или только участок У-хромосомы, содержащий генный локус. Он активизирует развитие гонад. У больных женщин не дифференцируются вторичные половые признаки, и они бесплодны.
Открытие
Впервые Х-хромосома была особенной в 1890 году Германом Хенкингом в Лейпциге. Хенкинг изучал яички пиррокориса и заметил, что одна хромосома не участвует в мейозе . Хромосомы названы так из-за их способности принимать окрашивание ( по-гречески цветность означает цвет ). Хотя Х-хромосома могла быть окрашена так же хорошо, как и другие, Хенкинг не был уверен, является ли это объектом другого класса, и поэтому назвал ее Х-элементом , который позже стал Х-хромосомой после того, как было установлено, что это действительно хромосома.
Идея о том, что Х-хромосома была названа из-за ее сходства с буквой «Х», ошибочна. Все хромосомы обычно выглядят под микроскопом как аморфные капли и принимают четко очерченную форму только во время митоза. Эта форма неопределенно X-образная для всех хромосом. Совершенно случайно, что Y-хромосома во время митоза имеет две очень короткие ветви, которые под микроскопом могут выглядеть слитыми и выглядеть как нисходящие элементы Y-образной формы.
Впервые было высказано предположение, что Х-хромосома участвует в определении пола Кларенсом Эрвином МакКлангом в 1901 году. Сравнив свою работу по саранче с Хенкингом и другими, Мак-Клунг отметил, что только половина сперматозоидов получила Х-хромосому. Он назвал эту хромосому дополнительной хромосомой и настаивал (правильно), что это правильная хромосома, и предположил (ошибочно), что это хромосома, определяющая самцов.
История открытия синдрома
Синдром получил свое название в честь Гарри Клайнфельтера — врача, в 1942 году впервые описавшего клиническую картину болезни. Клайнфельтер с коллегами опубликовали отчет об обследовании 9 мужчин, объединенных общими симптомами, такими как слабое оволосение тела, евнухоидный тип телосложения, высокий рост и уменьшенные в размерах яички. Позднее, в 1956 г., генетики Планкетт и Барр (Е. R. Plankett, М. L. Barr) обнаружили у мужчин с синдромом Клайнфельтера тельца полового хроматина в ядрах клеток слизистой оболочки полости рта, а в 1959 году Полани и Форд (P. E. Polanyi, S. E. Ford) с сотрудниками показали, что у больных в хромосомном наборе имеется лишняя Х-хромосома.
Активные исследования данной патологии велись в 70‑х годах в США. Тогда всех новорожденных мальчиков подвергали кариотипированию, в результате чего удалось достоверно выявить распространенность и генетические особенности синдрома Клайнфельтера.
Любопытно, что мыши также могут иметь синдром трисомии по половым хромосомам XXY, что позволяет эффективно использовать их в качестве моделей для исследования синдрома Клайнфельтера.
Хромосомы для чего нужны. Хромосомы человека
Интересные факты из мира медицины порой преподносят нам удивительные сюрпризы. Например, знаете ли вы, что такое хромосомы, и как они влияют на человека ?
Предлагаем разобраться в этом вопросе, чтобы раз и навсегда расставить все точки над «i».
Рассматривая семейные фотографии, вы наверняка могли заметить, что члены одного родства похожи друг на друга: дети – на родителей, родители – на бабушек и дедушек. Это сходство передается от поколения к поколению с помощью удивительных механизмов генетической наследственности .
У всех живых организмов, от одноклеточных водорослей до африканских слонов, в ядре клетки находятся хромосомы – тонкие длинные нити, которые можно рассмотреть только в электронный микроскоп.
Хромосо́мы (др.-греч. χρῶμα — цвет и σῶμα — тело) — это нуклеопротеидные структуры в ядре клетки, в которых сосредоточена бо́льшая часть наследственной информации (генов). Они предназначены для хранения этой информации, ее реализации и передачи.
Сколько хромосом у человека
Еще в конце XIX века ученые выяснили, что число хромосом у разных видов не одинаково.
Например, у гороха 14 хромосом, у крысы – 42, а у человека – 46 (то есть 23 пары) . Отсюда возникает соблазн сделать вывод, что чем их больше – тем сложнее существо, обладающее ими. Однако на самом деле это совершенно не так.
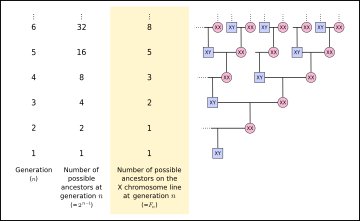
Из 23 пар человеческих хромосом 22 пары — аутосомы и одна пара — гоносомы (половые хромосомы). Половые имеют морфологические и структурные (состав генов) различия.
У женского организма пара гоносом содержит две Х-хромосомы (ХХ-пара), а у мужского – по одной Х- и Y-хромосоме (XY-пара).
Именно от того, каков будет состав хромосом двадцать третьей пары (ХХ или XY), зависит пол будущего ребенка. Определяется это при оплодотворении и слиянии женской и мужской половой клетки.
Данный факт может показаться странным, но по числу хромосом человек уступает многим животным. Например, у какой-то несчастной козы 60 хромосом, а у улитки – 80.
Хромосомы состоят из белка и молекулы ДНК (дезоксирибонуклеиновой кислоты), похожей на двойную спираль. В каждой клетке находится около 2 метров ДНК, а всего в клетках нашего организма около 100 млрд. км ДНК.
Интересен факт, что при наличии лишней хромосомы или при отсутствии хотя бы одной из 46, — у человека наблюдается мутация и серьезные отклонения в развитии (болезнь Дауна и т.п.).
Именно в связи с этим научным фактом в народе распространилось «интеллигентное» оскорбление: «У тебя что, лишняя хромосома?».
Теперь вы знаете общую информацию о принципах наследственности. Вообще эта тема очень интересная, хоть и чрезвычайно сложна.
Кстати, вас также может заинтересовать статья о том, почему близнецы похожи друг на друга .
Если вам нравятся интересные факты – подписывайтесь на InteresnyeFakty.org в любой социальной сети. С нами всегда интересно!
Что такое микроделеционный синдром?
Самые незначительные изменения (они же мутации) называются точечными. Их появление влияет на считанные единицы генов. В некоторых случаях нарушение относится вообще к одному единственному гену
Однако если он обеспечивал выработку важного белка, последствия для всего организма могут быть очень серьезными. Подобные патологические изменения относятся к группе микроделеционных синдромов
Каждое такое заболевание обусловлено небольшим изменением генетического материала, которое происходит в строго определенном месте. Точный механизм возникновения подобных нарушений на сегодняшний день не установлен, что не мешает ученым заниматься исследованием их воздействия на организм.
Так, было выяснено, что развитие синдрома в таком случае может происходить несколькими различными вариантами. В частности, ряд заболеваний характеризуется участием онкогенов. В других случаях на воздействие непосредственно самой делеции накладывается эффект хромосомного импринтинга и возможные однородительские дисомии.
Частота возникновения большей части микроделеционных синдромов крайне невелика: порядка 1 случая на 50-100 тысяч новорожденных. Набор клинических признаков обычно выражается отчетливо. Для того чтобы поставить диагноз, бывает достаточно лишь совокупности симптомов. Однако при таком подходе невозможно точно прогнозировать здоровье потомков, поэтому зачастую наряду с проверкой обычных признаков производится молекулярно-генетическая диагностика пробанда и его родственников (обычно родители, в некоторых случаях также требуется анализ генотипа братьев, сестер, теть, дядь и так далее).
Патологические проявления сильно отличаются. В частности, их проявление определяется тем, насколько большой участок генетического материала был утрачен в результате делеции. Кроме того, в ряде случаев играет роль то, от кого из родителей была получена мутация (влияние хромосомного импринтинга). Хорошей иллюстрацией последней ситуации является пара синдромов Прадера-Вилли и Ангельмана. Они оба обусловлены наличием делеции в 15 хромосоме. Однако из-за различного механизма действия при передаче от разных родителей клиническая картина этих заболеваний значительно отличается.
Аномалии генетического материала
Наследственный материал состоит из огромного количества нуклеотидов, формирующих гены. При этом в каждом гене последовательность нуклеотидов строго определена, поскольку должна кодировать определенный белок. Кроме того, сами гены при формировании хромосом также выстраиваются в фиксированном порядке. Благодаря сохранению этого порядка организм может функционировать, а ученые – быстро и точно указывать друг другу, про какой ген идет речь.
В идеальном случае система работает без малейших сбоев, а генетическая информация всегда передается в неизменном виде. Однако на практике большое число структурных единиц и постоянное воздействие различных факторов (например, ионизирующего излучения) приводит к тому, что время от времени возникают различные аномалии. В частности, отдельные участки последовательности ДНК могут быть скопированы на новое место. В таком случае говорят о дупликации. Если же вместо создания новой копии была перемещена часть исходной цепочки, то модификация называется транслокацией. Кроме того, иногда часть последовательности просто теряется, удаляется из генетического материала. В таком случае изменение называется делецией.
Поскольку взаимодействия в организме оттачивались в течение многих тысячелетий эволюционного развития, получилась очень слаженная система. И аномалии, даже самые небольшие, могут вызвать нарушение баланса. В таком случае в организме развивается то или иное нарушение. Если при этом причина находится на уровне генов, то говорят о генных болезнях. Если была утрачена или наоборот получена лишняя копия хромосомы, то такие нарушения называются хромосомными заболеваниями.