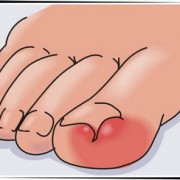
Диагноз «мозаичный синдром дауна»
Содержание:
Заболевания и расстройства
Следующие заболевания и расстройства связаны с генами на хромосоме 11:
- аутизм (нейрексин 1)
- острая перемежающаяся порфирия
- альбинизм
- атаксия – телеангиэктазия
- Синдром Беквита – Видеманна
- Болезнь Беста
- дефицит бета-кетотиолазы
- бета-талассемия
- Рак мочевого пузыря
- рак молочной железы
- дефицит карнитин пальмитоилтрансферазы I
- Болезнь Шарко – Мари – Зуба
- Муковисцидоз
- Депрессия
- Синдром Дениса-Драша
- семейная средиземноморская лихорадка
- Синдром Якобсена
- Синдром Джервелла и Ланге-Нильсена
- Лимфома из клеток мантии (t11; 14)
- Синдром Меккеля
- метгемоглобинемия , бета-глобиновый тип
- Смешанный лейкоз
- множественная эндокринная неоплазия 1 типа
- Наследственные множественные экзостозы
- Болезнь Ниманна – Пика
- несиндромная глухота
- порфирия
- Синдром Потоцкого-Шаффера
- Синдром Романо-Уорда
- Серповидноклеточная анемия
- Синдром Смита – Лемли – Опица
- дефицит тетрагидробиоптерина
- Синдром Ашера
- Синдром WAGR
- Синдром Видеманна – Штайнера
- Опухоль Вильмса
Заболевания и расстройства
Следующие заболевания связаны с генами на хромосоме 7:
- аргинино-янтарная ацидурия
- кавернозная мальформация головного мозга
- Болезнь Шарко – Мари – Зуба
- Холестаз, прогрессирующий семейный внутрипеченочный 3
- Цитруллинемия, тип II, у взрослых ,
- врожденное двустороннее отсутствие семявыносящего протока
- муковисцидоз
- Вербальная диспраксия, связанная с развитием
- дистальная спинномозговая мышечная атрофия V типа
- Синдром Элерса-Данлоса
- гемохроматоз, тип 3
- Наследственный неполипозный колоректальный рак HNPCC4
- Синдром лиссэнцефалии по типу нормана-робертса
- Синдром Марфана
- кленовый сироп болезнь мочи
- зрелый диабет у молодых людей 3-го типа
- мукополисахаридоз типа VII или синдром Слая
- Мышечная дистрофия, пояснично-конечностный тип 1D
- миелодиспластический синдром
- Врожденная миотония
- несиндромная глухота
- несовершенный остеогенез
- p47-фокс-дефицитная хроническая гранулематозная болезнь
- Синдром Пендреда
- Синдром Романо-Уорда
- Синдром Швахмана-Даймонда
- Шизофрения
- Синдром Сильвера-Рассела
- Специфические языковые нарушения
- Тританопия или тританомалия дальтонизм
- Синдром Вильямса
- Синдром Зеллвегера
Причины
45 хромосом у человека синдром Тернера вызван частичной или полной потерей (моносомой) одной из Х-хромосом.
Причина, по которой это происходит, неизвестна и считается результатом случайного события. В некоторых случаях хромосомная аномалия возникает спонтанно из-за ошибки разделения репродуктивных клеток родителя. Это приводит к тому, что генетическая ошибка содержится во всех клетках организма.
Во многих случаях затронут только определенный процент генов. Это называется мозаицизмом. Некоторые клетки имеют нормальные 46 хромосом (одна клеточная линия), в то время как другие не имеют их (вторая клеточная линия).
Эта вторая линия может содержать различные аномалии, такие как частичная или полная потеря Х-хромосомы. В этих случаях потеря генетического материала из Х-хромосомы обычно происходит из-за спонтанных ошибок во время раннего развития плода.
Более редкие аномалии хромосом (кроме полной или частичной моносомии) могут вызывать синдром Шерешевского Тернера. Такие аномалии включают кольцевую хромосому или изохромосому X.
В редких случаях некоторые клетки имеют одну копию Х-хромосомы, другие имеют одну копию Х и некоторый материал Y. Количество материала Y-хромосомы недостаточно, чтобы вызвать развитие мужских признаков, но связано с повышенным риском развития формы рака -гонадобластомы.
Большинство симптомов происходит из-за потери конкретного генетического материала из одной Х-хромосомы. Один ген убедительно доказал свою роль в развитии расстройства. SHOX кодирует белок, помогающий регулировать другие гены организма.
Белковый продукт гена SHOX играет роль в росте и созревании скелета. Исследователи считают, что потеря одного SHOX-гена на измененной Х-хромосоме является основной причиной дефицита роста у женщин с синдромом Тернера.
Общие вопросы
Интенсивное развитие генетики в течение последних десятилетий позволило развить отдельное направление хромосомной патологии, которая постепенно приобретает все большое значение. К этой области относятся не только хромосомные болезни, но и различные нарушения во время внутриутробного развития (к примеру, выкидыши). В настоящее время счет аномалий идет уже на 1000. Свыше ста форм характеризуются клинически очерченной картиной и называются синдромами.
Выделяется несколько групп болезней. Триплоидией называется случай, при котором в клетках организма имеется лишняя копия генома. Если же появился дубликат только одной хромосомы, то подобное заболевание называется трисомией. Также причинами аномального развития организма могут быть делеции (удаленные участки генетического кода), дупликации (соответственно, лишние копии генов или их групп) и иные дефекты. Английский врач Л. Даун в 1866 году описал одну из самых известных болезней такого рода. Синдром, получивший его имя, развивается при наличии лишней копии 21 хромосомы (трисомия-21). Трисомии по другим хромосомам, как правило, заканчиваются выкидышами или приводят к смерти в детском возрасте из-за серьезных нарушений в развитии.
Позже были открыты случаи моносомии по X-хромосоме. В 1925 году Шерешевский Н.А и в 1938 году Тернер Г. описали его симптомы. Трисомия-XXY, которая встречается у мужчин, была описана Клайнфельтером в 1942 году.
Указанные случаи заболеваний стали первыми объектами исследований в этой области. После того, как расшифровали этиологию трех перечисленных синдромов, фактически появилось направление хромосомных болезней. В течение 60-х годов дальнейшие цитогенетические исследования привели к формированию клинической цитогенетики. Ученые доказали связь между патологическими отклонениями и хромосомными мутациями, а также получили статистические данные о частоте появления мутаций у новорожденных и в случаях самопроизвольного прерывания беременности.
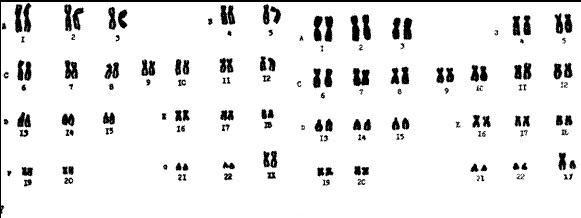
Типы хромосом. Принципы классификации хромосом. Денверская и Парижская классификации хромосом, их сущность.
Метафазная
хромосома состоит
из двух продольных нитей ДНП – хроматид,
соединенных друг с другом в области
первичной перетяжки (центромеры).
Центромера делит тело хромосомы на два
плеча. В зависимости от расположения
центромеры различают следующие типы
хромосом:
акроцентрические – центромера смещена к одному концу
хромосомы и одно плечо очень короткое;
субметацентрические –
центромера смещена от середины
хромосомы, и плечи имеют разную длину;
метацентрические –
центромера расположена посередине, и
плечи примерно одинаковой длины.
Участок
каждого плеча вблизи центромеры
называется – проксимальным, удаленный
от нее –дистальным. Концевые отделы
дистальных участков называются
теломерами. Теломеры препятствуют соединению концевых
участков хромосом. При потере этих
участков наблюдаются хромосомные
перестройки. Некоторые хромосомы могут
иметь вторичные
перетяжки ,
отделяющие от тел хромосомы участок,
называемый спутником .
Правила
хромосом.
Правило
постоянства числа хромосом.
Правило
парности хромосом.
Правило
индивидуальности хромосом.
Правило
непрерывности хромосом.
Денверская
классификация хромосом, которая
помимо размеров хромосом, учитывает их
форму, положение центромеры и наличие
вторичных перетяжек и спутников. 23 пары
хромосом человека разбили на 7 групп от
A
до G.
Важным параметром является центромерный
индекс (ЦИ), который отражает отношение
(в %) длины короткого плеча к длине всей
хромосомы.
К группе A относят 1-3 хромосомы. Это большие
метацентрические и субметацентрические
хромосомы, их центромерный индекс от
38-49.
Группа B (4 и 5 пары). Это большие субметацентрические
хромосомы, ЦИ 24-30.
Группа C (6-12 пары). Хромосомы среднего размера,
субметацентрические, ЦИ 27-35. К этой
группе относят и Х-хромосому.
Группа D (13-15 пары). Хромосомы акроцентрические,
сильно отличаются от всех других хромосом
человека, ЦИ около 15.
Группа E (16-18 пары). Относительно короткие,
метацентрические или субметацентрические,
ЦИ 26-40.
Группа F (19-20 пары): две короткие, субметацентрические
хромосомы, ЦИ 36-46.
Группа G (21-22 пары): это маленькие акроцентрические
хромосомы, ЦИ 13-33. К этой группе относят
и Y-хромосому.
В
основе Парижской
классификации хромосом человека (1971 г.) лежат методы специальной
дифференциальной их окраски, при которой
каждой хромосоме выявляется характерный
только для нее порядок чередования
поперечных светлых и темных сегментов.
Различные
типы сегментов обозначают по методам,
с помощью которых они выявляются наиболее
отчетливо (Q-сегменты, G-сегменты,
Т-сегменты, S-сегменты). Каждая хромосома
человека содержит свойственную только
ей последовательность полос, что
позволяет идентифицировать каждую
хромосому. Хромосомы спирализованы
максимально в метафазе, менее спирализованы
в профазе и прометафазе, что позволяет
выделить большее число сегментов, чем
в метафазе.
На
метафазной хромосоме (рис. 59) приводятся
символы, которыми принято обозначать
короткое и длинное плечо, а также
расположение районов и сегментов. В
настоящее время существуют ДНК-маркеры
или зонды, с помощью которых можно
определить изменение определенного,
даже очень маленького, сегмента в
хромосомах (цитогенетические карты).
На международном конгрессе генетики
человека в Париже в 1971 г. (Парижская
конференция по стандартизации и
номенклатуре хромосом человека) была
согласована система символов для более
краткого и однозначного обозначения
кариотипов. При
описании кариотипа :
•
указывается общее
число хромосом и набор половых хромосом ,
между ними ставится запятая (46, XX; 46, XY);
• отмечается какая хромосома лишняя или какой не
хватает (это указывается ее номером 5, 6 и др., или
буквами данной группы А, В и др.); знаком
«+» указывают на увеличение количества
хромосом , знаком
«-» указывают на отсутствие
данной хромосомы 47, XY,+ 21;
•
плечо хромосомы, в котором произошло
изменение ( удлинение
короткого плеча указывается символом ( р+); укорочение
(р-); удлинение
длинного плеча указывается символом
(q+); укорочение (q-); •
символы перестроек ( транслокация
обозначается t, а делеция — del )
помещают перед номерами вовлеченных
хромосом, а перестроечные хромосомы
заключают в скобки. Наличие двух
структурно-аномальных хромосом
обозначается точкой с запятой (;) или
нормальной дробью (15/21).
Половой хроматин в судебно-медицинском отношении
Исследование П. х. в суд.-мед. практике производится с целью установления половой принадлежности следов крови, слюны и других биол, жидкостей, вырванных волос, следов-отпечатков клеток тканей и органов, кусочков тканей, которые могут быть обнаружены на месте происшествия, на различных предметах, одежде, теле потерпевшего и подозреваемого в совершении преступления, на орудиях травмы, на транспортных средствах, а также при обнаружении обгоревших трупов или частей расчлененных трупов. Реже П. х. исследуют с целью суд.-мед. установления генетического пола у лиц с аномалиями полового развития, используя общепринятые методики.
Для приготовления препаратов из следов крови (см.) и слюны (см.) кусочки предмета-носителя помещают в пробирку и заливают 0,5—40% (следы крови) или 5—10% (следы слюны) уксусной к-той. Экстрагируют при комнатной температуре в течение нескольких часов и, удалив кусочки предмета-носителя, центрифугируют. Осадок переносят на предметное стекло и высушивают на воздухе. С пятен крови на предметах, не впитывающих жидкость (металл, стекло, пластмасса и др.), делают соскобы, которые затем обрабатывают таким же образом.
При исследовании волос (см.) корень волоса помещают на предметное стекло и добавляют 10—25% уксусную к-ту. После набухания отделяют и измельчают волосяной фолликул, удаляя остальные части волоса.
Из кусочков тканей и органов, при необходимости предварительно выдержав их до набухания в уксусной к-те соответствующей концентрации или в физиол, р-ре, готовят гистологические препараты, мазки или препараты-отпечатки. Следы-наложения клеток, тканей или органов на орудиях травмы смывают физиол, р-ром, одновременно соскабливая их. Мелкие кусочки тканей, встречающиеся в таких следах, измельчают препаровальными иглами. Смывы-соскобы помещают в пробирки, центрифугируют, из осадка готовят гистологические препараты. Исследование препаратов целесообразно начинать с выявления Y-хроматина, т. к. при его отсутствии те же препараты могут быть снова использованы для выявления X-хроматина. При исследовании учитывают только достаточно хорошо сохранившиеся неповрежденные ядра клеток. При анализе следов крови Y-хроматин определяют в ядрах лимфоцитов, т. к. в нейтрофилах Y-хроматин в препаратах из следов крови мужчин может не выявляться.
При отсутствии повышенной влажности Половой хроматин может длительно сохраняться в высохших следах, а также в клетках фолликула вырванного волоса. Высокая температура (выше 150°) разрушает ядра клеток и П. х. Значительная влажность в течение нескольких суток также приводит к разрушению клеток, что делает невозможным выявление полового хроматина. Т. к. условия, в к-рых находятся следы, могут последовательно меняться, решающее значение для установления пригодности следов крови, слюны и т. д. для определения П. х. имеет состояние обнаруживаемых в них клеток и их ядер. В клетках высохших кусочков тканей, не подвергающихся действию влаги, П. х. сохраняется длительное время. В целых трупах и в их крупных частях в процессе аутолиза и гниения в течение нескольких суток происходит деструкция клеточных ядер. В обгоревших трупах половой хроматин нек-рое время может сохраняться в клетках глубоко расположенных органов и тканей.
При выявлении небольшого числа клеток, сохранивших ядра, исследуемых на Половой хроматин, с целью установления статистической достоверности результатов используют различные математические методы анализа, учитывающие как общее число обнаруженных клеток, так и число клеток. содержащих X- или Y-хроматин.
Библиография: Давиденкова Е. Ф., Берлинская Д. К. и Тысячнюк С. Ф. Клинические синдромы при аномалиях половых хромосом, Л., 1973; Захаров А. Ф. Хромосомы человека, М., 1977; Капустин А. В. Судебно-медицинская диагностика пола по половым различиям в клетках, М., 1969; Лабораторные и специальные методы исследования в судебной медицине, под ред. В. И. Пашковой и В. В. Томилина, с. 157, М., 1975; Любинская С. И. и Антонова С. Н. Исследование Y-хроматина в следах крови, Суд.-мед. экспертиза, т. 18, № 3, с. 17, 1975; Основы цитогенетики человека, под ред. A. А. Прокофьевой-Бельговской, М., 1969; Methods in human cytogenetics, ed. by H. G. Schwarzacher a. U. Wolf, p. 207, B. а. о., 1974; The sex chromatin, ed. by K. L. Moore, Philadelphia — L., 1966.
Исследование хромосомных отклонений
Первые исследования эффектов от хромосомных нарушений стали проводить в 60-х годах, после того как был установлен хромосомный характер некоторых заболеваний. Можно условно выделить две большие группы связанных эффектов: врожденные пороки развития и изменения, вызывающие летальные исходы. Современная наука располагает сведениями, что хромосомные аномалии начинают проявляться уже на стадии зиготы. Летальные эффекты при этом являются одной из основных причин гибели плода в утробе (этот показатель у человека достаточно высок).
Хромосомные аберрации – это изменение структуры хромосомного материала. Они могут как возникать спорадически, так и передаваться по наследству. Точная причина, по которой они появляются, не установлена. Ученые полагают, что за некоторую часть таких мутаций отвечают различные факторы окружающей среды (например, химически активные вещества), которые воздействуют на эмбрион или даже на зиготу. Интересен тот факт, что большая часть хромосомных аберраций обычно связана с хромосомами, которые зародыш получает от отца.
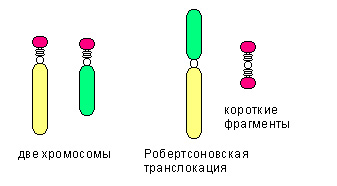
Значительная часть хромосомных аберраций встречается очень редко и была обнаружена один раз. В то же время некоторые другие достаточно часто встречаются, причем даже у людей, не связанных родственными узами. К примеру, широко распространена транслокация центромерных или близких к ним районов 13 и 14 хромосом. Утрата неактивного хроматина коротких плеч практически не влияет на состояние здоровья. При аналогичных робертсоновских транслокациях в кариотип попадает 45 хромосом.
Примерно две трети всех обнаруживаемых у новорожденных хромосомных аномалий компенсируются за счет других копий генов. По этой причине они не несут серьезной угрозы нормальному развитию ребенка. Если же компенсация нарушения невозможна, возникают пороки развития. Часто такая несбалансированная аномалия выявляется у больных с умственной отсталостью и другими врожденными пороками, а также у плода после самопроизвольных абортов.
Известны компенсированные аномалии, которые способны наследоваться из поколения в поколение без возникновения заболеваний. В некоторых случаях такая аномалия может перейти в несбалансированную форму. Так, если имеется транслокация, затрагивающая 21 хромосому, возрастает риск трисомии по ней. По статистике такие транслокации имеются у каждого 20 ребенка, у которого зафиксирована трисомия-21, причем в каждом пятом случае аналогичное нарушение есть у одного из родителей. Поскольку большая часть детей с вызванной транслокацией трисомией-21 рождается у молодых (менее 30 лет) мам, то в случае обнаружения этого заболевания у ребенка необходимо произвести диагностическое обследование молодых родителей.
Риск появления нарушений, которые не компенсируются, сильно зависит от транслокации, поэтому теоретические расчеты затруднены. Тем не менее, приблизительно определить вероятность соответствующей патологии можно на основании статистических данных. Такая информация собрана для распространенных транслокаций. В частности, робертсоновская транслокация между 14 и 21 хромосомами у матери с вероятностью 2 процента приводит к трисомии-21 у ребенка. Эта же транслокация у отца передается по наследству с вероятностью 10%.
Помоги своему ребенку
Мозаичный вариант СД, несмотря на более легкое течение, требует постоянной работы с такими детьми. К сожалению, окончательно избавиться от патологии невозможно, но с помощью многопрофильной терапии можно помочь ребенку лучше адаптироваться к социуму и не чувствовать себя обделенным.
Очень важно для детей с синдромом Дауна проводить массаж и лечебную физкультуру. Они позволят повысить тонус мышц и укрепить их, устранят контрактуры, стабилизируют работу суставов и всего опорно-двигательного аппарата
Массаж показан с 2 недель рождения. Но не все приемы позволительно использовать в этом возрасте. Разминание не применяется детям до 3 месяцев. Если у малыша есть проблемы с сердцем, следует проконсультироваться с врачом по поводу массажных приемов.
Из методов ЛФК обязательно используйте упражнения на мяче. Благодаря им происходит развитие двигательных рефлексов и координации.
Показаны для детей с СД занятия плаванием. Оно выполняет функцию гидромассажа.
Особо выделяют такие методы лечения, как иппотерапия и дельфинотерапия.
Иппотерапия – это терапевтическое воздействие, достигаемое при общении с лошадьми. Такой метод обеспечивает:
- развитие равновесия и внимания;
- придает уверенности в себе;
- тепло, которое исходит от лошади, успокаивает ребенка и дает ему чувство защищенности.
Это отличный метод, позволяющий уравновесить нервную систему людей с любым типом синдрома Дауна. Но он требует выполнения некоторых условий:
Важно, чтобы лошадь была большая, а не пони. Они чрезмерно суетливы.
Перед тем как сесть на лошадь, с ней нужно установить контакт: поговорить, покормить, погладить.
Животное должно быть без седла и подков.
Не рекомендуется кататься по асфальту
Дельфинотерапия обеспечивает расслабление и улучшение настроения, придает уверенности в себе, помогает приобрести навыки общения. Этот метод также стимулирует физическое развитие. Ультразвук, который выделяют дельфины, восстанавливает нормальные биотоки.
К другим способам терапии относят:
- назначение медикаментозных средств — гормоны, психостимуляторы, нейрометаболиты, витаминные комплексы;
- консультации психолога и психотерапевта;
- диетотерапия — нередко такие больные страдают ожирением, которое способно приводить к другим патологиям. Поэтому следует соблюдать систему правильного питания, чтобы предотвратить чрезмерный набор веса;
- постоянное наблюдение у узких специалистов.
Изменчивость хромосом в онтогенезе и эволюции
Постоянство числа хромосом в хромосомном наборе и структуры каждой хромосомы — непременное условие нормального развития в онтогенезе (см.) и сохранения биол. вида. В течение жизни организма могут происходить изменения числа отдельных хромосом и даже их гаплоидных наборов (геномные мутации) или структуры хромосом (хромосомные мутации). Необычные варианты хромосом, обусловливающие уникальность хромосомного набора индивидуума, применяются в качестве генетических маркеров (маркерных хромосом). Геномные и хромосомные мутации играют важную роль в эволюции биол. видов. Данные, полученные при изучении хромосом, вносят большой вклад в систематику видов (кариосистематику). У животных одним из главных механизмов эволюционной изменчивости является изменение числа и структуры отдельных хромосом
Важное значение имеет также изменение содержания гетерохроматина в отдельных или нескольких хромосомах. Сравнительное изучение хромосом человека и современных человекообразных обезьян позволило на основании сходства и различия индивидуальных хромосом установить степень филогенетического родства этих видов и смоделировать кариотип их общего ближайшего предка.
Библиогр.:
Бочков Н. П., Захаров А. Ф. и Иванов В. И. Медицинская генетика, М., 1984; Дарлингтон С. Д. и Ла Кур Л. Ф. Хромосомы, Методы работы, пер. с англ., М., 1980, библиогр.; Захаров А. Ф. Хромосомы человека (проблемы линейной организации;, М., 1977, библиогр.; Захаров А. Ф. и др. Хромосомы человека, Атлас, М., 1982; Кикнадзе И. И. Функциональная организация хромосом, Л., 1972, библиогр.; Основы цитогенетики человека, под ред. А. А. Прокофьевой-Бельговской, М., 1969: Суонсо н К., Мерц Т. и Янг У. Цитогенетика, пер. с англ., М., 1969; Cell biology, A comprehensive treatise, ed. by L. Goldstein a. D. M. Prescott, p. 267, N. Y. a. o., 1979; Seuanez H. N, The phylogeny of human chromosomes, v. 2, B. a. o. 1979; Sharm a A. K. a. Sharma A. Chromosome techniques, L. a. o., 1980; ThermanE. Human chromosomes, N. Y. a. o., 1980.
Дети с хромосомной патологией
Дети с хромосомной патологией имеют определенные внешние признаки. Синдром Дауна характеризуется косыми глазными щелями, плоской переносицей, плоским профилем лица. Плоский профиль лица встречается почти у 90% детей с синдромом Дауна, плоская переносица встречается у 65% больных детей. Дети с хромосомной патологией, синдромом Дауна, имеют отличительные черты – открытый рот, слегка высунутый язык, эпикант, характерный низкий рост волос на затылке, также на затылке отмечается излишняя кожа. Эти признаки патологии встречаются в 80% случаев синдрома Дауна, в 60% случаев отмечаются диспластические уши, короткие пальцы, узкое нёбо. У ребенка с синдромом Дауна изменяется форма зубов – они приобретают вид острых клыков, изменяется внешний вид языка – язык напоминает географический рельеф, ему присваивают название – «географический язык». Синдрому Дауна сопутствуют многие нарушения развития — умственная отсталость, мышечная гипотония, которая встречается в 80% случаев патологии. Патология развития сердца при синдроме Дауна выявляется в среднем у 50% больных детей. Дети с хромосомной патологией синдром Дауна имеют сниженный иммунитет.
Хромосомная патология синдром Дауна имеет несколько форм:
- Простая форма — хромосомная патология синдром Дауна, хромосома 47.ХХ. 21+. Хромосомная патология простой формы встречается часто — в 95% случаев синдрома Дауна.
- Мозаичная форма — хромосомная патология синдром Дауна, хромосома 47. ХY.21+/46. ХY, встречается редко, в 1% случаев патологии.
- Транслокационная форма — хромосомная патология синдром Дауна, хромосома 47.ХХ.t 21|15; а также 47. XY/t 21/21, встречается примерно в 4% случаев этой патологии. В случае Робертсоновской транслокации возможно рождение у носителей генетической транслокации,ребенка с синдромом Дауна:
- 45.ХХ.t 21/15 (мать) – от 10 до 15%.
- 45.ХY.t 21/15 (отец) – от 5 до 7%.
- 45.ХY.t 21/21 (любой из родителей) – 100%.
Дети с синдромом Дауна, должны проходить стимуляцию центральной нервной системы – специфическую и неспецифическую, хирургическое лечение, если оно показано. Дети с синдромом Дауна, как правило, очень послушны и исполнительны. При правильном воспитании они могут ухаживать за собой, ухаживать за домашними животными, хорошо читать, петь, полностью повторять действия взрослого во время выполнения работ. Дети с хромосомной патологией должны проходить социальную реабилитацию для адаптации в обществе, специальное обучение, при достижении определенного возраста — посильное трудоустройство.
Аномалии генетического материала

Наследственный материал состоит из огромного количества нуклеотидов, формирующих гены. При этом в каждом гене последовательность нуклеотидов строго определена, поскольку должна кодировать определенный белок. Кроме того, сами гены при формировании хромосом также выстраиваются в фиксированном порядке. Благодаря сохранению этого порядка организм может функционировать, а ученые – быстро и точно указывать друг другу, про какой ген идет речь.
В идеальном случае система работает без малейших сбоев, а генетическая информация всегда передается в неизменном виде. Однако на практике большое число структурных единиц и постоянное воздействие различных факторов (например, ионизирующего излучения) приводит к тому, что время от времени возникают различные аномалии. В частности, отдельные участки последовательности ДНК могут быть скопированы на новое место. В таком случае говорят о дупликации. Если же вместо создания новой копии была перемещена часть исходной цепочки, то модификация называется транслокацией. Кроме того, иногда часть последовательности просто теряется, удаляется из генетического материала. В таком случае изменение называется делецией.
Поскольку взаимодействия в организме оттачивались в течение многих тысячелетий эволюционного развития, получилась очень слаженная система. И аномалии, даже самые небольшие, могут вызвать нарушение баланса. В таком случае в организме развивается то или иное нарушение. Если при этом причина находится на уровне генов, то говорят о генных болезнях. Если была утрачена или наоборот получена лишняя копия хромосомы, то такие нарушения называются хромосомными заболеваниями.