Саркома легких
Содержание:
Диагностика саркомы костей
·Для верификации онкопроцесса поразившего кость и определения дальнейшей тактики лечения, необходимо определить гистологический тип опухоли. “Золотым стандартом” — является выполнение трепан-биопсии либо открытой биопсии.
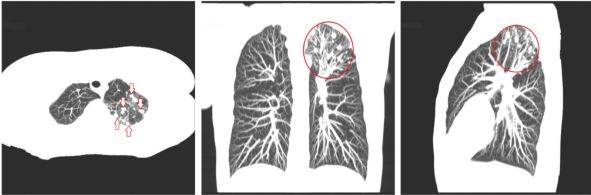
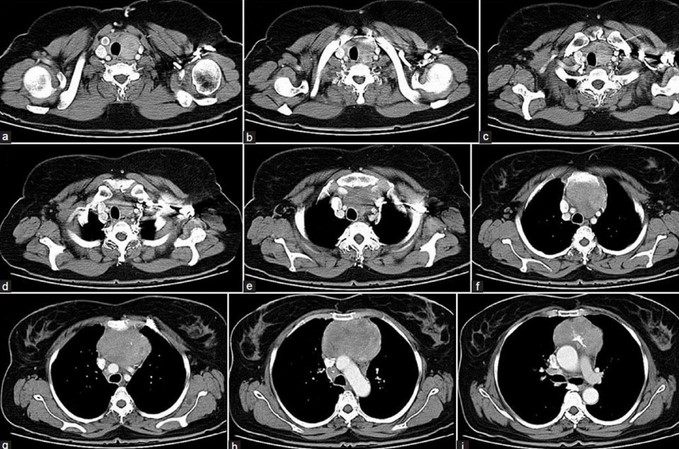
·Компьютерная томография легких. Наиболее часто саркомы костей метастазируют в легкие, а разряжающая возможность флюорографии и рентгеновского исследования невелика, поэтому пациентам с саркомами костей необходимо выполнение компьютерной томографии органов грудной клетки.
·Компьютерная томография или магнитно-резонансная томография первичного очага опухоли. Эти исследования позволяют адекватно оценить распространенность опухолевого процесса, прилежание опухоли к магистральным сосудисто-нервным структурам, степень вовлечения прилежащих мягких тканей.
·Ультразвуковое исследование брюшной полости, печени, прилежащих зон регионарного лимфооттока. Саркомы костей редко метастазируют в печень и регионарные лимфотические коллекторы (3-5%) и наличие метастазов в них является неблагоприятным прогностическим фактором, однако проводить исследование этих областей так же необходимо.
·Сцинтиграфия костей. Второй по частоте развития метастазов сарком костей точкой являются кости, поэтому проведение сцинтиграфии для таких пациентов обязательно.
Лечение саркомы костей
В первую очередь следует рассматривать возможность выполнения органосохранных операций. Главным условием операбельности пациентов является радикальность и абластичность удаления опухоли, что гарантирует отсутствие местного рецидива.
Сотрудники ФГБУ «НМИЦ радиологии» Минздрава России владеют всеми современными методиками органосохранной хирургии опорно – двигательного аппарата:
· Эндопротезирование онкологическим эндопротезом плечевого сустава.
· Эндопротезирование онкологическим эндопротезом тазобедренного сустава.
· Эндопротезирование онкологическим эндопротезом коленного сустава.
· Тотальное эндопротезирование бедренной кости.
· Модульное эндопротезирование вертлужной впадины после удаления опухолей костей таза.
· Эндопротезирование тел позвонков на всех уровнях позвоночного столба.
· Эндопротезирование онкологическим эндопротезом лучезапястного сустава.
· Эндопротезирование онкологическим эндопротезом локтового сустава.
· Реконструкция удаленных фрагментов костей при помощи ауто/аллопластики.
· Резекция костей плечевого пояса (лопатка, ключица).
· Резекции ребер и грудной стенки.
· Использование собственной кости пациента для замещения дефекта после операции.
· Вертебропластика.
· Онкологическое эндопротезирование, металлолстеосинтез.
· Расширенные операции при массивных опухолях плечевого пояса (межлопаточно-грудная ампутация).
При невозможности выполнения органосохранных операций необходимо выполнение калечащих операций в радикальном объеме. Ампутация или экзартикуляция конечности выполняются в следующих случаях:
- обширное первично-множественное распространение опухоли;
- вовлечение в опухолевый процесс магистрального сосудисто-нервного пучка.
- технически исключающий проведение реконструктивно-пластического этапа операции;
- прогрессирование на фоне предоперационной химиотерапии;
- отказ пациента от органосохранной операции;
- жизненные показания к экстренной хирургии – распад опухоли, кровотечения.
Симптомы Саркомы (ангиосаркомы) Капоши:
Чаще всего болезнь проявляется поражением кожи, но способна затрагивать и слизистые оболочки, и лимфатическую систему, и внутренние органы (прежде всего легкие и ЖКТ). Все формы саркомы Капоши обусловлены инфекцией, вызываемой герпесвирусом человека типа 8, который передается половым путем, с кровью или слюной. За несколько месяцев до появления очагов саркомы Капоши в результате присутствия герпесвируса человека типа 8 в крови образуются специфические антитела. Недавно описаны случаи возникновения кожных очагов, по времени совпадающие с сероконверсией Классификация саркомы Капоши:
Классический тип
- Эндемический тип
- Эпидемический тип
- Иммунно-супрессивный тип
Классический тип
Распространён в Центральной Европе, России и Италии. Излюбленные локализации саркомы Капоши классического типа — это стопы, боковые поверхности голени, поверхности кистей. Очень редко на слизистых оболочках и веках. Очаги поражения обычно симметричны, асимптомны, но редко могут быть зуд и жжение. Границы очагов, как правило, чёткие. Различают 3 клинические стадии:
- пятнистая
- папулезная
- розеолезная
Пятнистая. Самая ранняя стадия. Пятна на этой стадии красновато-синюшного или красновато-бурого цвета диаметром от 1 мм до 5 мм, неправильной формы. поверхность гладкая. Папулезная. Элементы в эту стадию сферической или полусферической формы, плотно-эластической консистенции, от 2 мм до 1 см в диаметре. Чаще изолированные. При слиянии образуют бляшки уплощенной или полушаровидной формы. Поверхность бляшек гладкая или шероховатая (по типу апельсиновых корок). Опухолевая. На этой стадии происходит образование единичных или множественных узлов. Диаметром от 1-5 см, красно-синюшного или синюшно-бурого цвета. Мягкие или плотно-эластической консистенции, сливающиеся и изъязвляющиеся. Эндемический тип
Распространён, в основном, у жителей Центральной Африки. Начинается преимущественно в детском возрасте, пик заболеваемости приходится на первый год жизни ребёнка. Как правило, поражаются внутренние органы и главные лимфоузлы. Кожные поражения редки и минимальны. Эпидемический тип
Эта саркома Капоши, ассоциированная со СПИДом, является наиболее достоверным симптомом ВИЧ-инфекции. Характерен молодой (до 37 лет) возраст, яркость окраски и сочность высыпных элементов. Необычна и локализация опухолей: на кончике носа и слизистых оболочках, на твердом небе и верхних конечностях. Отмечается быстрое течение заболевания с обязательным вовлечением лимфоузлов и внутренних органов. Иммунно-супрессивный тип
Протекает, как правило, хронически и доброкачественно. Развивается после пересадки почки, после которой назначаются особые типы иммунносупрессоров. При отмене препаратов наступает регресс заболевания. Внутренние органы вовлекаются редко. Выделяют острую, подострую и хроническую формы саркомы Капоши. Острую форму саркомы Капоши отличает быстрая генерализация процессса. Нарастающие симптомы общей интоксикации и кахексия служат причиной смерти в течение 2 мес-2 лет. Подострая форма саркомы Капоши имеет менее быстрое и не столь злокачественное течение. Длиительность болезни без лечения может достигать 2-3 лет. Относительно доброкачественным течением, постепенным прогрессированием процесса отличается хроническая форма саркомы Капоши, при которой длиительность заболевания может составлять 8-10 лет и более. Осложнения саркомы Капоши определяются стадией болезни и локализацией очагов поражения. Возможны ограничения движения в конечностях, их деформация, кровотечения и интоксикация при распадах опухолей, нарушения зрения при соответствующей локализации очагов поражения и др.
Что провоцирует / Причины Саркомы Юинга:
Для костных сарком характерен быстрый рост и раннее метастазирование. Саркома Юинга является второй по частоте среди злокачественных опухолей костей у детей — составляет 10-15 %. Эта опухоль редко встречается у детей моложе 5 лет и у взрослых старше 30 лет. Пик заболеваемости приходится на 10−15 лет.
Причина появления злокачественных опухолей костей пока не известна, однако доказано, что в 40 % возникновение костной саркомы провоцирует травма.
Существует некоторая связь между возникновением саркомы Юинга и наличием скелетных аномалий (энхондрома, аневризмальная костная киста и т.д.) и аномалиями мочеполовой системы (гипоспадии, редупликация почечной системы). В отличие от остеосаркомы, ионизирующая иррадиация не ассоциируется с возникновением саркомы Юинга.
Цитогенетический анализ показывает в 85% случаев хромосомную транслокацию t(11,22) (q24, q12) в большинстве клеток, выделенных из этой опухоли. Аналогичные изменения выявляются в другой мелкоклеточной опухоли — PNET (примитивной нейроэктодермальной опухоли). И хотя саркома Юинга не имеет анатомической связи со структурами ЦНС или автономной симпатической нервной системы, эти цитогенетические изменения доказывают нейроэктодермальную природу опухоли. Кроме того, в большинстве случаев в клетках опухоли выявляется экспрессия РАХ3 протеина, который в норме определяется в период эмбрионального развития нейроэктодермальной ткани. При саркоме Юинга часто можно определить и другой опухолевый маркер — NSE (сывороточная нейрон-специфичная энолаза).
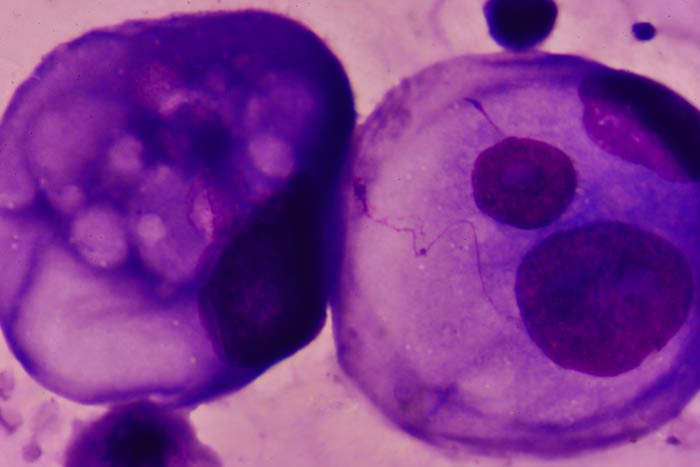
Саркома Юинга состоит из мелких круглых клеток со скудной цитоплазмой, круглым ядром, содержащим нежный хроматин и слабо просматривающиеся базофильные нуклеолы. В отличие от остеосаркомы, она не продуцирует остеоид.
Идея эндотелиальной природы опухоли Юинга превалировала до 1980 года. Исследования, проведенные в последние годы, показали нейрогенную природу опухоли Юинга. Хотя чаще саркома Юинга является недифференцированной опухолью костей, имеются данные о поражении мягких тканей (экстраоссальная саркома Юинга). В специальной литературе появился термин «семейство опухолей типа саркомы Юинга».
Факторы риска развития саркомы Юинга
В настоящее время известно несколько факторов риска, связанных с возникновением саркомы Юинга.
Пол. Саркома Юинга несколько чаще встречается среди мальчиков по сравнению с девочками.
Возраст. В 64% случаев саркома Юинга встречается в возрасте от 10 до 20 лет.
Раса. Наиболее часто саркома Юинга наблюдается у белого населения.
Развитие заболевания
На саркому могут указывать:
- боль в костях в ночное время, которая не проходит после приема стандартных доз анальгетиков;
- образование припухлости, которая быстро увеличивается в размерах, сначала безболезненная, затем появляется болевой синдром.
Рост опухоли затрагивает соседние органы и ткани, что приводит к появлению соответствующих симптомов. Например, при поражении нервных стволов болевые ощущения передаются по ветви нервной системы, а распространение опухоли на трахею ведет к проблемам с дыханием.
Паростальная саркома костной ткани и некоторые другие виды заболевания могут протекать бессимптомно годами, однако в большинстве случаев опухоль развивается быстро. Оно прогрессирует в течение нескольких месяцев или недель и характеризуется обширным распространением с ранним образованием метастазов.
Такие заболевания, как липосарком, могут проявляться в нескольких местах организма последовательно или одновременно.
Течение заболевания во время беременности
Лимфосаркома может сочетаться с беременностью. Возможны следующие варианты:
- Зачатие наступило в состоянии ремиссии.
- Опухоль была выявлена во время внутриутробного развития плода.
- Развился рецидив новообразования в период беременности.
Беременность и роды в период стойкой ремиссии не отягощают прогноза опухоли. Однако риск рецидивирования лимфосаркомы гораздо выше для тех пациенток, у которых зачатие происходит в течение первых 2-3 лет после полной стабилизации состояния. В связи с этим женщины должны быть предупреждены о необходимости применения контрацептивов в течение этого периода.
Лимфосаркома высокого риска, которая была диагностирована в первом триместре, служит показанием к прерыванию беременности. При обнаружении опухоли во втором и третьем триместре тактика ведения подбирается индивидуально, с привлечением таких специалистов, как онколог, неонатолог, терапевт, генетик. Учитывается степень распространенности заболевания и характер ответа на лечение.
При рецидиве опухоли прогноз для беременности самый неблагоприятный. В данной ситуации требуется применение высокотоксичных препаратов, прием которых несовместим с беременностью.
Диагностика леймиосаркомы
Своевременно проведенная диагностика повышает шансы на полное выздоровление и предотвращает метастазирование опухоли. Для выявления заболевания применяются следующие методики:
- биопсия опухолевых тканей: если леймиосаркома локализуется во внутренних органах, то для забора материала используют пункционные или эндоскопические методы;
- гистологическое исследование забранного материала во время биопсии;
- ультразвуковая диагностика;
- рентгеновское исследование;
- магнито-резонансная, компьютерная томография.
Диагностику злокачественных новообразований необходимо проводить как можно раньше, в специализированных диагностических центрах. Расшифровкой полученных результатов должны заниматься опытные специалисты, знакомые с особенностями развития леймиосаркомы и их отображением на снимках.
Что провоцирует / Причины Остеогенной саркомы:
Развитие опухоли имеет некоторую связь с быстрым ростом кости. Дети, страдающие остеосаркомой, как правило, выше ростом, по сравнению с возрастной нормой, и болезнь поражает наиболее быстро растущие части скелета
Развитие костных опухолей часто ассоциируется с травмой, но, скорее травма привлекает внимание врача и заставляет провести рентгенологическое исследование
Единственный агент внешней среды, известный как стимулятор костных сарком — ионизирующее излучение. Причем интервал между воздействием этого фактора и возникновением остеосаркомы может быть от 4 до 40 лет (в среднем 12 — 16 лет)
Среди страдающих болезнью Педжета 2% заболевают остеосаркомой, часто с множественным поражением костей.
Наличие доброкачественных опухолей костей (остеохондромы, энхондромы и т.д.) увеличивает риск заболевания остеосаркомой.
Среди пациентов, излеченных от ретинобластомы, 50% вторичных опухолей приходится на остеосаркому (ретинобластома — опухоль, часто имеющая наследственный характер), и при обоих заболеваниях встречаются одинаковые изменения в 13 паре хромосом.
Какие шансы вылечиться от саркомы/опухоли мягких тканей?
Шансы детей и подростков вылечиться от саркомы мягких тканей или от редкого вида опухоли мягких тканей зависят от нескольких причин. Самые важные из них — это конкретный вид опухоли, её размер, насколько она успела вырасти в организме к моменту диагноза, можно ли удалить опухоль хирургически, а также возраст заболевшего ребёнка.
Благодаря тому, что в последние десятилетия детей с саркомами мягких тканей или с редкими видами опухоли мягких тканей лечат по стандартным протоколам , результаты эффективности лечения значительно выросли. Если ещё в 70-ые годы выживало только от 30 до 40 % заболевших детей, то сегодня 10-летняя выживаемость составляет в среднем около 70%. Прогресса удалось добиться благодаря тому, что подходы к лечению болезни постоянно корректируются в зависимости от результатов исследований.
Если стечение разных причин у ребёнка складывается благоприятно, то результаты долговременной выживаемости могут превышать и 80%. Но если к моменту диагноза у ребёнка опухоль очень большого размера и её невозможно удалить, болезнь уже успеоа перейти на и/или опухоль уже успела дать метастазы в другие части тела, то такая ситуация является неблагоприятной. А вместе с ней снижаются шансы на долговоременное выживание, как говорят медики.
Необходимое замечание: названные проценты выздоровевших являются статистическими показателями. Они точно и достоверно описывают лишь совокупность заболевших саркомой/опухолью мягких тканей. Статистика не может предсказать, выздоровеет конкретный ребёнок, или нет. Какой конкретно прогноз болезни у Вашего ребёнка, спрашивайте у лечащего врача.
Патогенез (что происходит?) во время Саркомы (ангиосаркомы) Капоши:
Мультицентрическое развитие, медленная эволюция идиопатического типа саркомы Капоши, возможность регресса очагов поражения, а также наличие гистологических признаков воспаления при отсутствии признаков клеточной атипии позволяют предполагать, что хотя бы в начале своего развития саркома Капоши является, скорее всего, реактивным процессом, а не истинной саркомой. Гистогенез саркомы Капоши не ясен
Противоречивы и мнения о происхождении веретенообразных клеток — важного компонента бляшек и опухолей при саркоме Капоши. Предположение о том, что они являются трансформированными эндотелиальными клетками, было подтвержено результатами ультраструкгурньгх и иммуногистохимических исследований с маркерами CD31 и С34
Существует мнение о более вероятном происхождении саркомы Капоши из эндотелия лимфатических, а не кровеносных капилляров. Что касается гипотезы о развитии этих клеток из дермальныхдендроцитов, она не нашла подтверждения в исследованиях с использованием маркеров XI Па фактора против маркеров факторов von Willebrand VIII, несмотря на присутствие вокруг узлов опухоли реактивной гиперплазии дендроцитов. Тем не менее существует предположение, что активированные дендроциты могут играть важную роль в инициации очага саркомы Капоши. В последние годы проводятся молекулярнобиологические исследования факторов промоции при развитии саркомы Капоши. При экспериментальном изучении характерного для саркомы Капоши неоангиогенеза в культуре клеток опухоли были выделены цитокины, стимулирующие рост клеточных культур данной опухоли, такие как интерлейкин 6 (IL-6), фактор роста фибробластов ((3FGF), трансформирующий фактор роста p(TGFp). Большое значение в развитии саркомы Капоши при СПИДе отводится онкостатину М-цитокину, вырабатываемому макрофагами и активированными Т-лимфоцитами. Он первично образуется в веретенообразных клетках саркомы капоши и является аутокринным фактором роста для СПИД-ассоциированной саркомы Капоши. Нейтрализация онкостатина М-специфическими олигонуклеотидами уменьшает рост клеточныхлиний саркомы Капоши и снижает продукцию IL-6. Механизм действия онкостатина М опосредован через тирозинкиназы, а его влияние может быть нейтрализовано ингибитором тирозинкиназ — генистеином. Хотя ВИЧ не считается этиологическим фактором саркомы Капоши, регуляторный генный продукт tat, имеющий ВИЧ-происхождение, может индуцировать развитие подобных саркоме Капоши очагов у мышей. Tat стимулирует in vitro рост веретенообразных клеток из предшественников сосудистых клеток. Такая стимуляция блокируется анти-1а1-антителами. Tat-протеин связывается с а5Ы и avb3 интегрин-рецепторами и усиливает связь эндотелиальных клеток. При исследовании онкогенов, связанных с саркомой Капоши, было показано, что веретенообразные клетки избыточно экспрессируют онкоген ras, в котором были определены точечные мутации . Int-2 — онкогенный продукт, также известный как FGF3, экспрессируется в 55% образцов саркомы Капоши. Обычно опухоль имеет пурпурную окраску, но цвет может иметь различные оттенки: красный, фиолетовый или бурый. Опухоль может быть плоской или слегка возвышаться над кожей, представляет собой безболезненные пятна или узелки. Почти всегда располагается на коже, реже — на внутренних органах. Саркома Капоши часто сочетается с повреждением слизистой нёба, лимфоузлов. Течение заболевания медленное. Обнаружение саркомы Капоши при ВИЧ-инфекции даёт основание для постановки диагноза СПИД. Гистологическая структура опухоли характеризуется множеством хаотично расположенных тонкостенных новообразованных сосудов и пучков веретенообразных клеток. Характерна инфильтрация опухоли лимфоцитами и макрофагами. Сосудистый характер опухоли резко увеличивает риск кровотечений. Однако делать биопсию при подозрении на саркому Капоши вовсе не обязательно. Саркома Капоши — особый вид опухоли, который часто не требует не только верификации диагноза, но и его лечения. Это может показаться странным на первый взгляд. Такая ситуация связана с тем, что поставить безошибочный диагноз можно и без биопсии, а изолированное лечение саркомы Капоши крайне редко даёт полное исцеление. Более того, лечение саркомы Капоши (в силу своей связи с причинными факторами основного заболевания) обычно является паллиативным, то есть направленным лишь на уменьшение симптомов заболевания.
Симптомы развития сарком мягких тканей
Патология характеризуется появлением безболезненного узелка или незначительной припухлости. Новообразование часто имеет четкие контуры, но если злокачественный очаг залегает глубоко, то края новообразования могут быть неровными и трудноопределимыми. Кожный покров над опухолью практически не отличается от здоровой ткани, может наблюдаться местное повышение температуры.
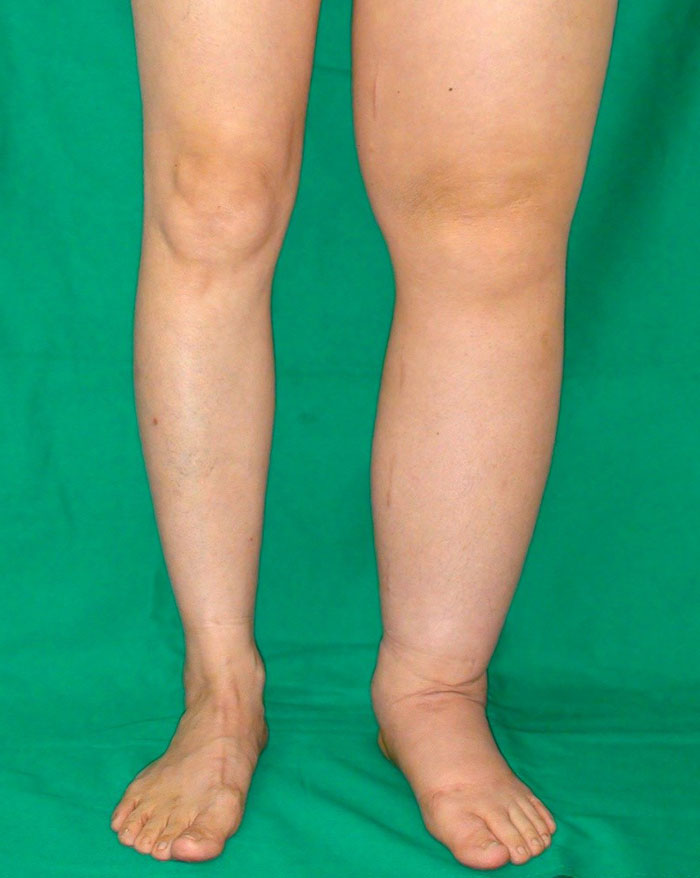
С увеличением роста новообразования часто возникает расширенная венозная сеть и язвенные участки. Во время движения могут возникать дискомфортные ощущения, хотя выраженного нарушения подвижности конечности при этом заболевании не наблюдается.
Список литературы:
-
Juergens C, Weston C, Lewis I, Whelan J, Paulussen M, Oberlin O, Michon J, Zoubek A, Jürgens H, Craft A: Safety assessment of intensive induction with vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) in the treatment of Ewing tumors in the EURO-E. W.I. N.G. 99 clinical trial. Pediatric blood & cancer 2006, 47: 22
-
Kaatsch P, Spix C: German Childhood Cancer Registry — Jahresbericht / Annual Report 2015 (1980-2014). Institut für Medizinische Biometrie, Epidemiologie und Informatik (IMBEI), Universitätsmedizin der Johannes Gutenberg-Universität Mainz 2015 [URI: http://www.kinderkrebsregister.de/ dkkr/ ergebnisse/ jahresbericht/ jahresbericht-2015.html]
-
Dirksen U, Jürgens H: Ewing-Sarkome des Kindes- und Jugendalters. S1-Leitlinie der Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH) AWMF online 2014 [URI: https://www.awmf.org/ uploads/ tx_szleitlinien/ 025-006l_S1_Ewing_Sarkome_Kinder_Jugendliche_2014-06-abgelaufen.pdf]
-
Haeusler J, Ranft A, Boelling T, Gosheger G, Braun-Munzinger G, Vieth V, Burdach S, van den Berg H, Jürgens H, Dirksen U: The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES). Cancer 2010, 116: 443
-
Burkhardt B, Reiter A, Landmann E, Lang P, Lassay L, Dickerhoff R, Lakomek M, Henze G, von Stackelberg A: Poor outcome for children and adolescents with progressive disease or relapse of lymphoblastic lymphoma: a report from the berlin-frankfurt-muenster group. Journal of clinical oncology 2009, 27: 3363
-
Wagner LM, McAllister N, Goldsby RE, Rausen AR, McNall-Knapp RY, McCarville MB, Albritton K: Temozolomide and intravenous irinotecan for treatment of advanced Ewing sarcoma. Pediatric blood & cancer 2007, 48: 132
-
Gerth HU, Juergens KU, Dirksen U, Gerss J, Schober O, Franzius C: Significant benefit of multimodal imaging: PET/CT compared with PET alone in staging and follow-up of patients with Ewing tumors. Journal of nuclear medicine : official publication, Society of Nuclear Medicine 2007, 48: 1932
-
Germeshausen M, Skokowa J, Ballmaier M, Zeidler C, Welte K: G-CSF receptor mutations in patients with congenital neutropenia. Current opinion in hematology 2008, 15: 332
-
Bernstein M, Kovar H, Paulussen M, Randall RL, Schuck A, Teot LA, Jürgens H: Ewing’s sarcoma family of tumors: current management. The oncologist 2006, 11: 503
-
Hunold A, Weddeling N, Paulussen M, Ranft A, Liebscher C, Jürgens H: Topotecan and cyclophosphamide in patients with refractory or relapsed Ewing tumors. Pediatric blood & cancer 2006, 47: 795
-
Hawkins DS, Schuetze SM, Butrynski JE, Rajendran JG, Vernon CB, Conrad EU 3rd, Eary JF: Fluorodeoxyglucose positron emission tomography predicts outcome for Ewing sarcoma family of tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2005, 23: 8828
-
Dirksen U, Poremba C, Schuck A: Knochentumoren des Kindes-und Jugendalters. Der Onkologe 2005, 10: 1034
-
Niemeyer C, Kontny U: Tumoren der Ewing-Sarkom-Familie, in: Gutjahr P (Hrsg.): Krebs bei Kindern und Jugendlichen. Deutscher Ärzte-Verlag Köln 5. Aufl. 2004, 491
-
Schuck A, Ahrens S, Paulussen M, Kuhlen M, Konemann S, Rube C, Winkelmann W, Kotz R, Dunst J, Willich N, Jürgens H: Local therapy in localized Ewing tumors: results of 1058 patients treated in the CESS 81, CESS 86, and EICESS 92 trials. Int J Radiat Oncol Biol Phys 2003, 55: 168
-
Paulussen M, Frohlich B, Jürgens H: Ewing tumour: Incidence, prognosis and treatment options. Paediatr Drugs 2001, 3: 899